
Why is everyone here lying?
— Fyodor Dostoevsky
Life requires carbohydrates, nucleic acids, lipids, and proteins. What is the chemistry behind their origin? Biologists seem to think that there are well-understood prebiotic molecular mechanisms for their synthesis. They have been grossly misinformed. And no wonder: few biologists have ever synthesized a complex molecule ab initio. If they need a molecule, they purchase molecular synthesis kits, which are, of course, designed by synthetic chemists, and which feature simplistic protocols.
Polysaccharides? Their origin?
The synthetic chemists do not have a pathway.
The biologists do not have a clue.
Making Molecules
Chemists study molecules.1 Synthetic chemists make them. What nature does is anyone’s guess. The molecules that we make are made to perform certain functions. The initial design is important. Sometimes molecular designs are computer-assisted, but more often than not, the initial steps are done on paper. A target must first be drawn or otherwise designated. This is no trivial task. In some cases, chemists have seen the target in a related system; in other cases, they guess the target’s properties on the basis of its molecular weight, its shape, its addends, and its functional capacities.
This is just the beginning.
Once a target is selected, retrosynthesis is next, whether on paper or on a computer screen. Placing the target at the top, the chemist draws an inverted tree (or graph), one step down at a time, into multiple branch points, until he reaches a level where starting materials are at hand.2
The decision tree is then pruned. Certain branches lead to dead ends. They are lopped off. Further refinement of various routes leads to a set of desired paths; these are the routes that can be attempted in the laboratory.
Why the retrosynthetic approach to complex molecules? It is because finding a direct path to a target is far too complicated. Dead ends are everywhere; dead products accumulate massively; and, between the dead ends and the dead products, precious starting materials tend to become exhausted.
There are no targets in evolution. Nature does not perform retrosynthetic analyses.
Given a target and a path to get there, the synthetic chemist must now try a number of chemical permutations. Each step may need to be optimized, and each step must be considered with respect to specific reaction site modifications and different reaction rates.
What is desired is often ever so slightly different in structure from what is not. If Product A is a mirror image of Product B, separation becomes a time-consuming and challenging task, one requiring complementary mirror-image structures. Many molecules in natural biological systems are homochiral. Their mirror images cannot do their work.
Few reactions ever afford a one hundred percent yield; few reactions are free of deleterious byproducts. Purification is essential. If byproducts are left in reaction, they result in complex mixtures that render further reactions impossible to execute correctly.
After purification, a number of different spectroscopic and spectrometric methods must be used to confirm the resulting molecular structures. Make the wrong molecular intermediate, the synthetic chemist quickly learns, and all subsequent steps are compromised.
Intermediate products are often unstable in air, sunlight or room light, or in water. Synthetic chemists work in seconds or minutes.
It is this laborious trench work that separates the men from the boys.
The Synthesis
Consider the synthesis of a molecular machine, a nanovehicle, a simple unimolecular structure that can translate itself along a surface when supplied with thermal or photonic energy. We have published before on these topics for the chemical literature, and this work is taken, in large part, often verbatim, from some of our synthetic papers on nanovehicles.3
Figure 1.
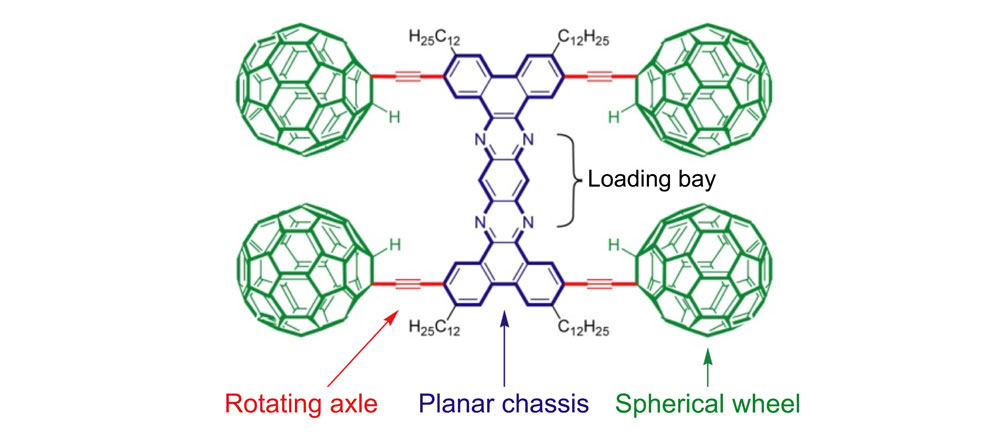
Figure 1a. Nanotruck 1a
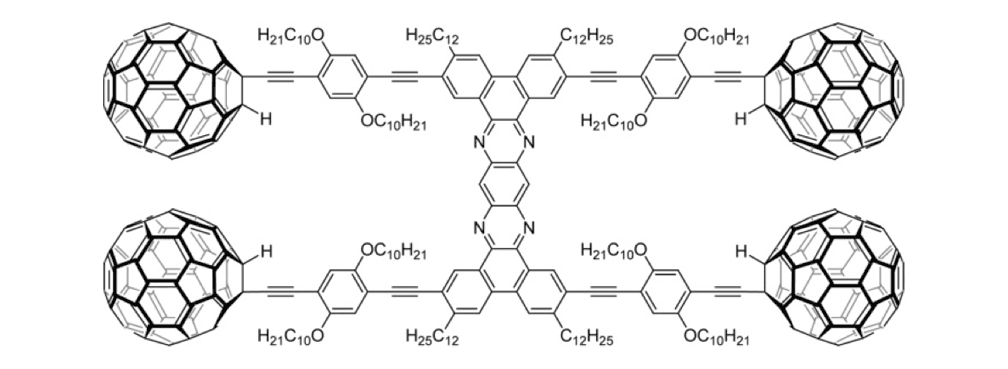
Figure 1b. Nanotruck 1b
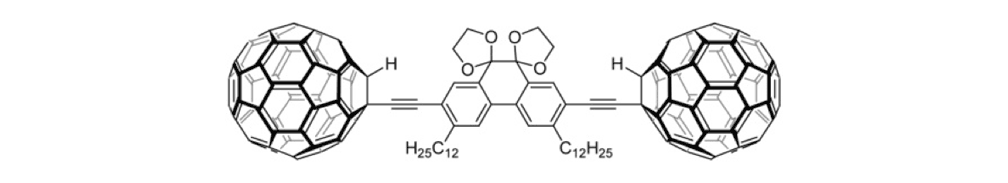
Figure 1c. Halftruck 2
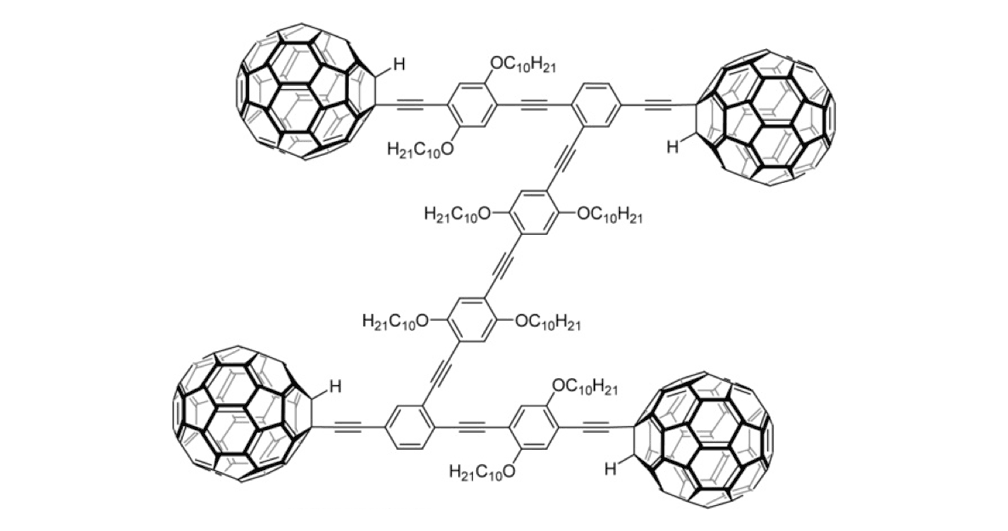
Figure 1d. Nanocar 3a
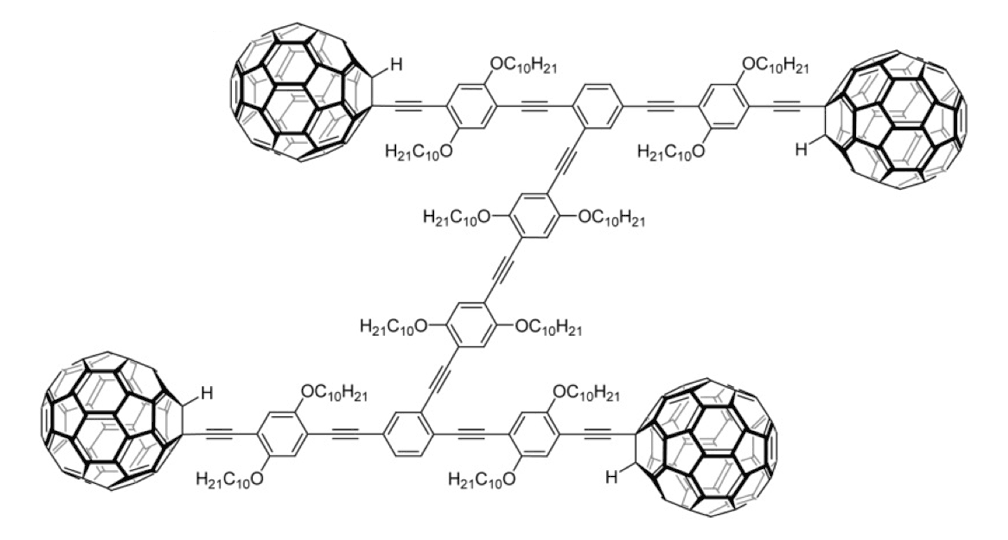
Figure 1e. Nanocar 3b
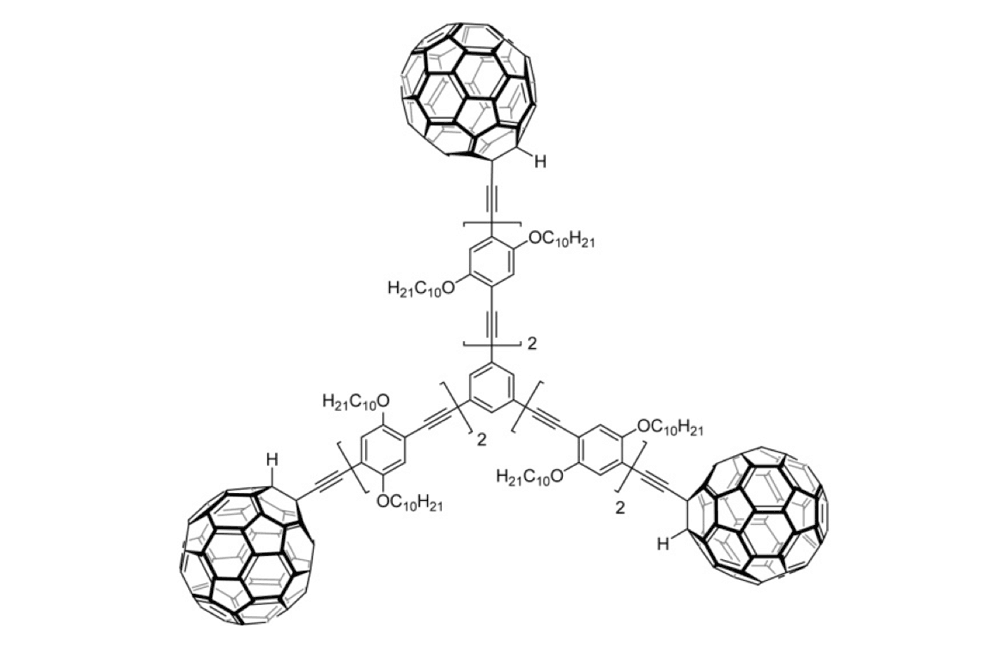
Figure 1f. Trimer 4a
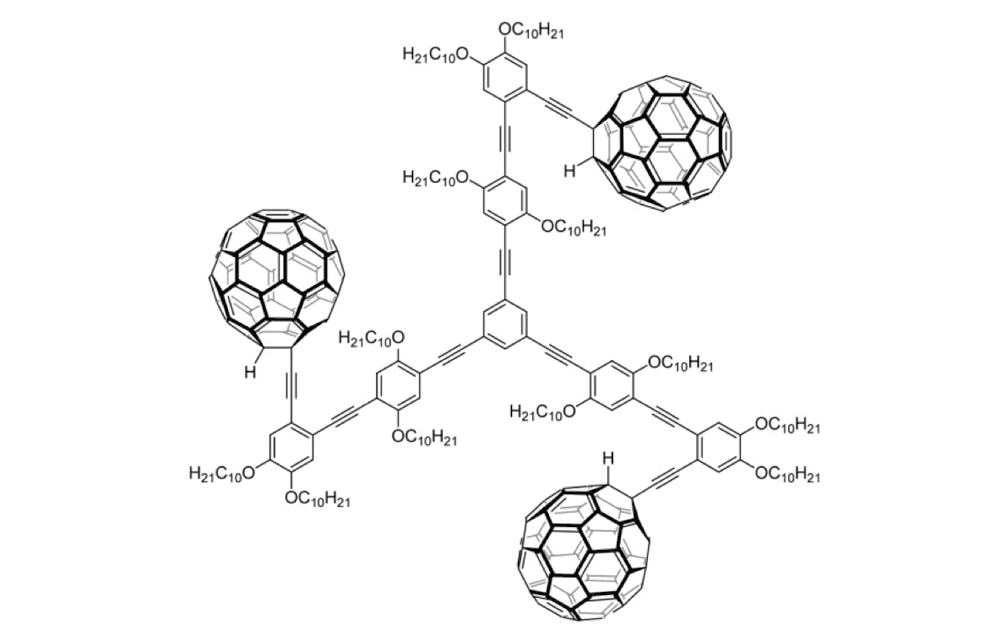
Figure 1g. Trimer 4b
The structures of the fullerene-wheeled molecular machines developed in the present study.
Figure 1a. Molecular structures of the molecular machines.
Nanocar 4a only rotates on axis; it does not translate as does nanocar 3b, and the difference between the two substantiates the wheel rotation.4
As shown in Figure 1a, nanotrucks and nanocars consist of three basic molecular mechanical parts: fullerene wheels, a chassis made of fused aromatic rings or oligo(phenylene ethynylene)s (OPEs), and alkynyl axles. The potential loading bays of nanotrucks 1a and 1b are based on carbon bonding to nitrogen atoms. Not so in nanocars 3a and 3b.
We developed a new methodology involving the in situ ethynylation of fullerenes to synthesize these molecular machines. The syntheses of several related molecular structures indicated that the alkyl units would be critical for the requisite solubility of multi-fullerenes.
Our nanomachines had spherical fullerene wheels that were connected to a chassis via freely rotating alkynyl axles. These highly flexible groups have minimal interactions with surfaces, at least in comparison to the strong charge transfer interaction of a fullerene wheel on gold. Ours was the first molecular-sized machine that incorporated mechanical components, such as wheels and axles, with movement at the single molecular level.5
The rolling motion of these nanocars resembled the rolling motion of macroscopic cars.
Using a fullerene wheel and alkynyl axle, we expected that the symmetrical spherical structure of C60 would allow the smooth rotation of a molecular wheel. Free rotation of the alkynyl axles between the fullerene wheel and chassis would then facilitate the rolling motion of the entire molecular vehicle on solid surfaces.
To check this design, we examined the rotational barrier of the wheel and axle architecture using ab initio methods at the HF/3-21G level of theory (Figure 2).6 This permits us to understand the molecular orbital interactions around the triple bond, which needs to rotate easily. Even though the phenylene-ethynylene and fullerene π-systems have been shown to exhibit periconjugation, its contribution to the rotational barrier was expected to be small since the largest estimated barrier height of the three model compounds was only about 1.0 kcal/mol. This height is actually comparable to that of the triple bond rotation in the usual phenylene-ethynylene system, allowing the free rotation of the fullerene-wheel structures at room temperature, or even under cryogenic conditions.
Figure 2.
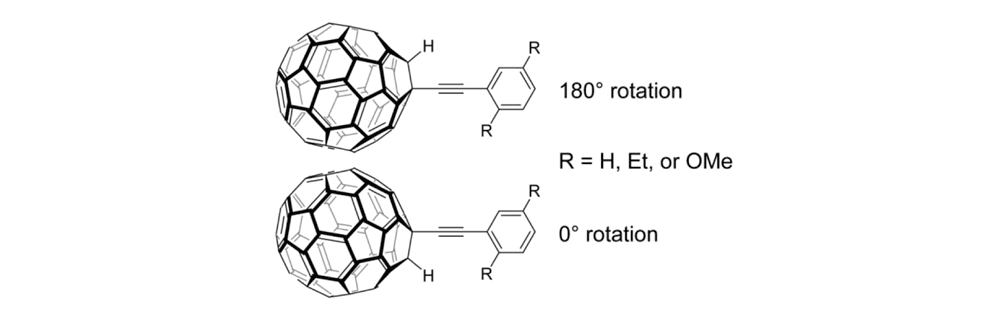
Figure 2a.

Figure 2b.
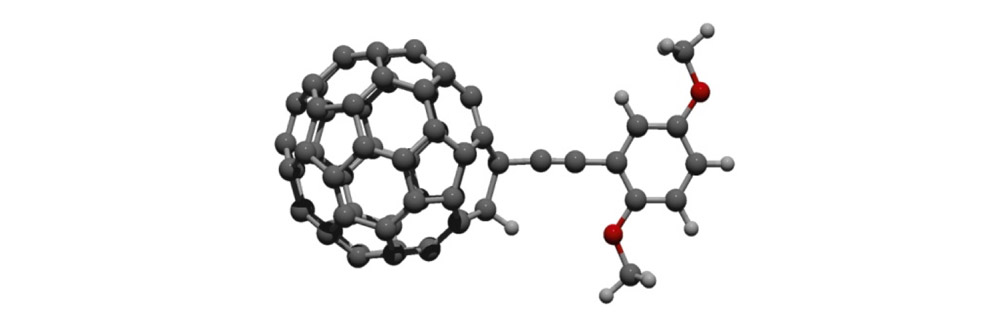
Figure 2c.
Rotational barrier of the fullerene-wheel structures. Using HF/3-21G level of theory on the fullerene-wheel model compounds with three different substituents (Figure 2a); relaxed potential energy surface scans around the wheel rotation were performed (Figure 2b) (the least stable conformation of each model was set to 0kcal/mol).
Figure 2c. Optimized structure of the fullerene-wheel model (R = OMe) at 0° rotation angle.7
Another key was chassis design. We chose two different structures: a rigid structure in nanotrucks 1a and 1b, and a semi-rigid structure in nanocars 3a and 3b. These fullerene-wheeled nanostructures shared several features.
First, most of the proposed structures weighed more than 4,000 daltons (Da) due to the high molecular weight of each C60 (720Da).
Second, a semi-rigid OPE chassis remedied difficulties encountered in the rigid chassis system, and enabled us to perform single molecule imaging studies by scanning tunneling microscopy (STM). Better flexibility of OPE structure combined with the increased number of alkyl units dramatically increased the solubility of the fullerene-wheeled structures in the organic solvents in which they were synthesized. The integration of as many as four C60s in a well-defined molecular system had not previously been achieved for a rigid molecular system because fullerene derivatives are so difficult to synthesize; this, in turn, made it difficult to obtain pure nanotruck samples.
Third, the rotation around alkyne connections in nanocars 3a and 3b between the chassis and axle portions in the OPE system can cause it to act as a suspension system; this gives the nanocar flexibility orthogonal to the surface plane (Figure 3), and permits it to roll onto gold domains that are one atomic step in height: pure gold.
Figure 3.
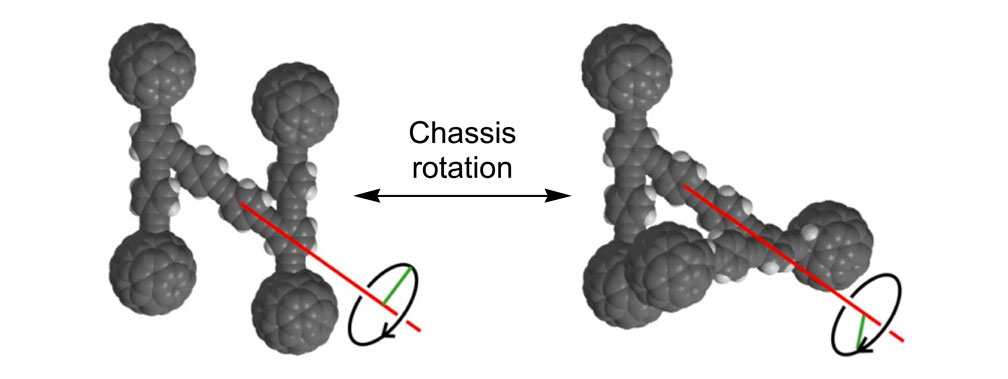
Figure 3a.
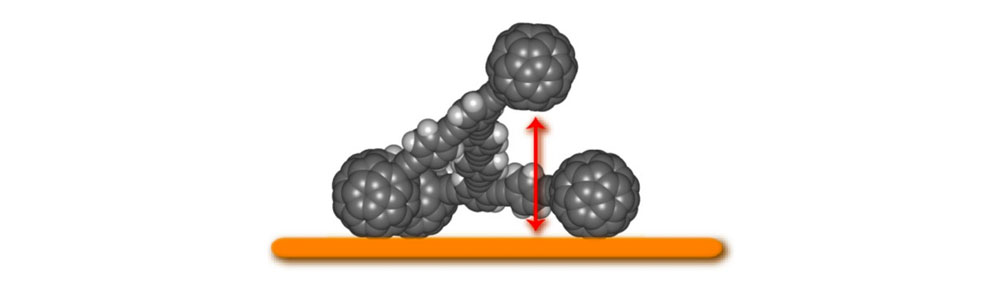
Figure 3b.
Flexibility of the semi-rigid chassis structure (nanocar 3a, devoid of alkoxy groups for clarity).
Figure 3a. The triple bonds in the OPE structure can rotate until the fullerene wheels touch one another, which gives the nanocar flexibility orthogonal to the surface plane.
Figure 3b. One fullerene wheel is elevated while the other wheels remain on the surface to illustrate the suspension concept.8
Half-Trucks
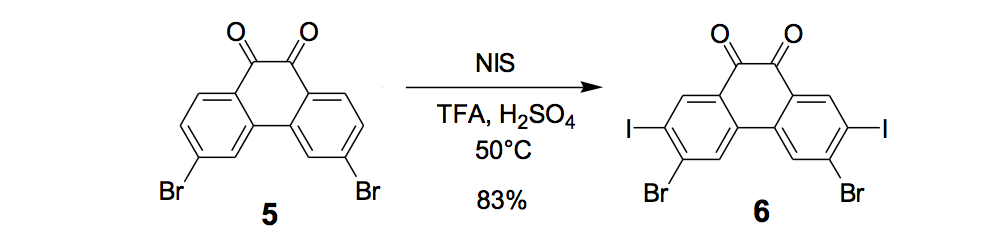
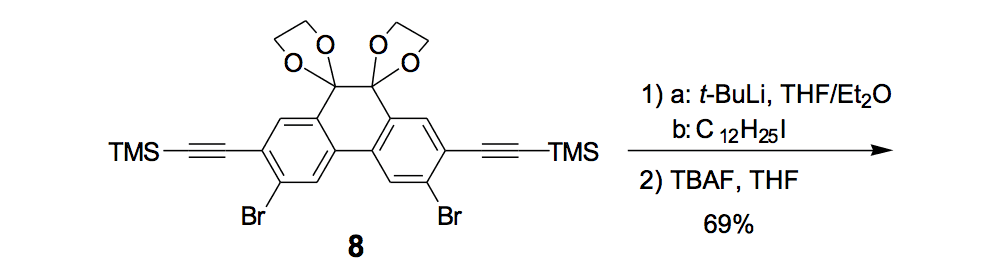
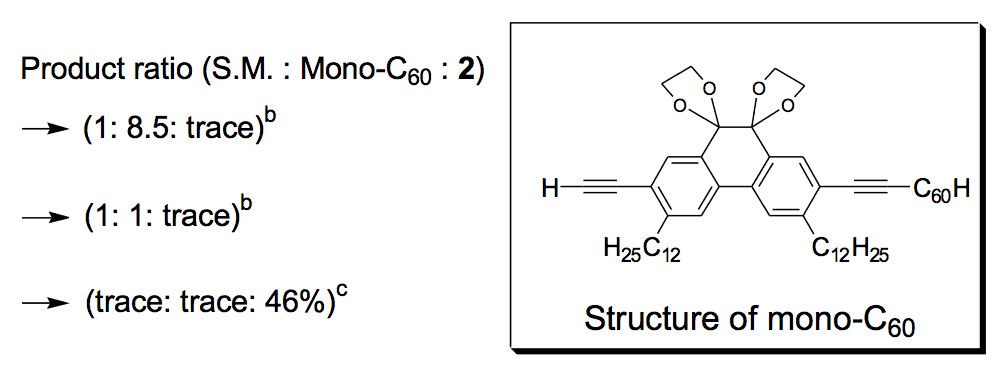
The synthesis of the prototype nanotruck, half-truck 2 (as illustrated in Figure 1c), is outlined below in Scheme 1. Half-truck 2 was designed and synthesized to develop a reliable method of attaching fullerenes to terminal alkynes. Dibromo-diketone 5 (Scheme 1) was iodinated to give compound 6. Protection of the diketone moiety of compound 6 was achieved with ethylene glycol. Compound 7 underwent a Pd-catalyzed coupling reaction with trimethylsilylacetylene (TMSA) to give compound 8. After the lithium-halogen exchange on the bromide followed by quenching with n-dodecyl iodide, the TMS protecting group was removed using tetrabutylammonium fluoride (TBAF) in THF to obtain compound 9 in 69% yield over two reaction steps. The key reaction step was achieved in 46% yield after testing under numerous different conditions.
The breakthrough was achieved when the terminal alkynes were deprotonated in THF using excess lithium hexamethyldisilazide (LHMDS) in the presence of excess C60.
Deviation from any one of the conditions dramatically affected reaction outcomes.
Scheme 1.
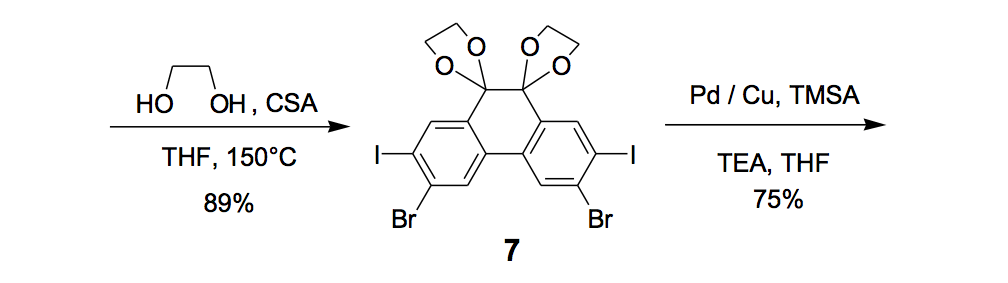

Synthesis of the prototype: half-truck.9
a Reagent: Pd / Cu = PdCl2(PPh3)2, CuI. b Yield determined by 1H NMR. c Isolated yield.
This in situ approach was applicable to other substrates, and resulted in the preparation of a wide variety of multi-fullerene structures. This methodology was so effective that the attachment of as many as four C60s was achieved on nanocars and nanotrucks. Eventually, all of our C60-wheeled nanotrucks and nanocars were synthesized by applying this methodology to tetra-terminal alkyne precursors.
The precision needed for this reaction in terms of temperature and reagent addition sequences accentuates the care needed to effect organic reactions.
Nanotrucks
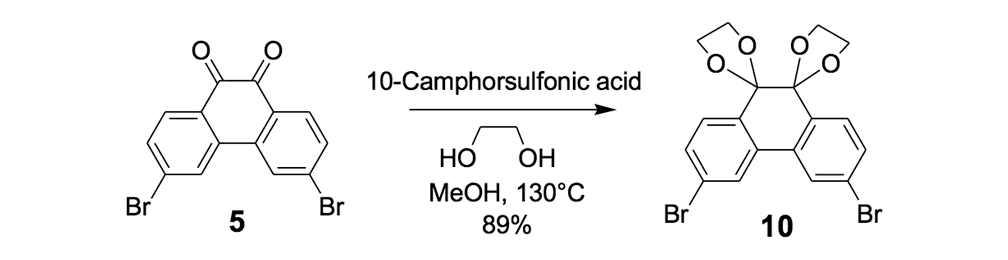
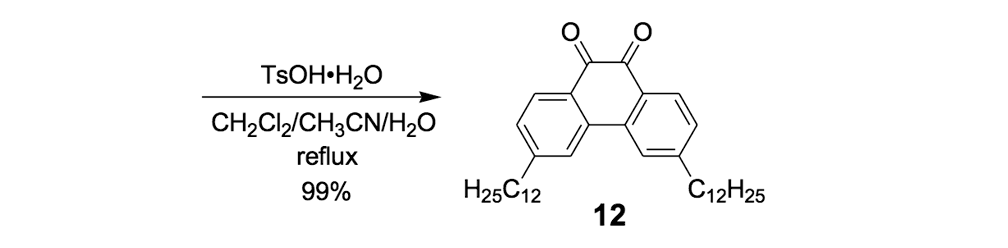
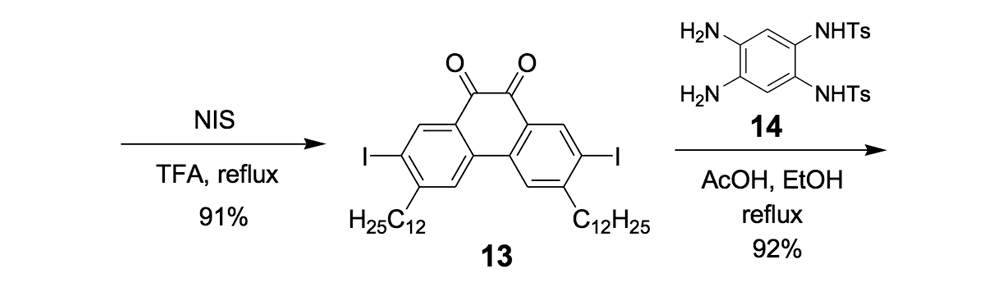
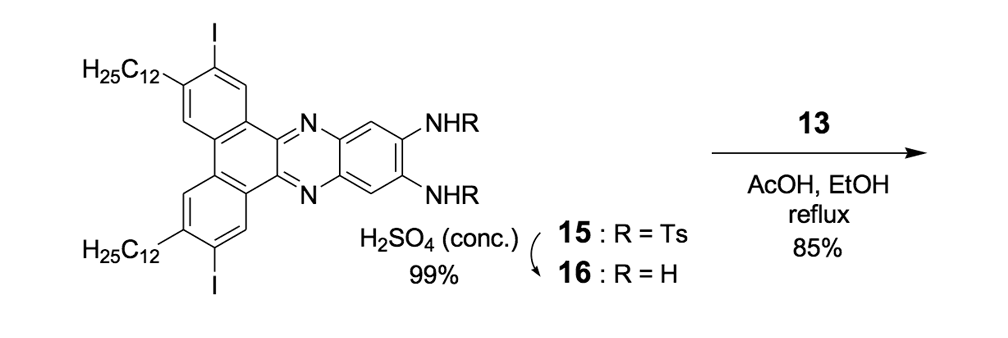
The syntheses of nanotrucks 1a and 1b are outlined below in Scheme 2. Half-truck 2, or its precursors, half-trucks 8 and 9 (Scheme 1), could not be used in the synthesis of nanotrucks 1a or 1b (Figures 1a and 1b) because deprotection of the diketone moiety with various aqueous acids was not possible in the presence of alkynes. Protecting 5 (Scheme 1) with ethylene glycol under CSA catalysis yielded diacetal 10 (Scheme 2). Compound 10 underwent a lithium-halogen exchange reaction with t-BuLi to form a dilithiated species directly alkylated by n-dodecyl iodide to yield compound 11. Deprotection of compound 11 with TsOH in water yielded diketone 12. Iodination of diketone 12 with NIS in trifluoroacetic acid (TFA) thus resulted in diiodide 13. Compound 13 was condensed with 14 (prepared by bis-tosylation of 1,2-phenylene diamine, 4,5-dinitration with HNO3, and finally reduction with Sn/HCl) to yield the half-chassis 15. Compound 15 had its protecting groups removed with H2SO4 to yield free diamine 16, which was subsequently condensed with compound 13 to form the full chassis 17. Compound 17 was then cross-coupled with TMSA under Pd-catalysis to make the chassis-axle component 18. Desilylation of 18 with KOH, followed by the in situ ethynylation in THF with excess C60 using LHMDS, yielded nanotruck 1a (Figure 1a).

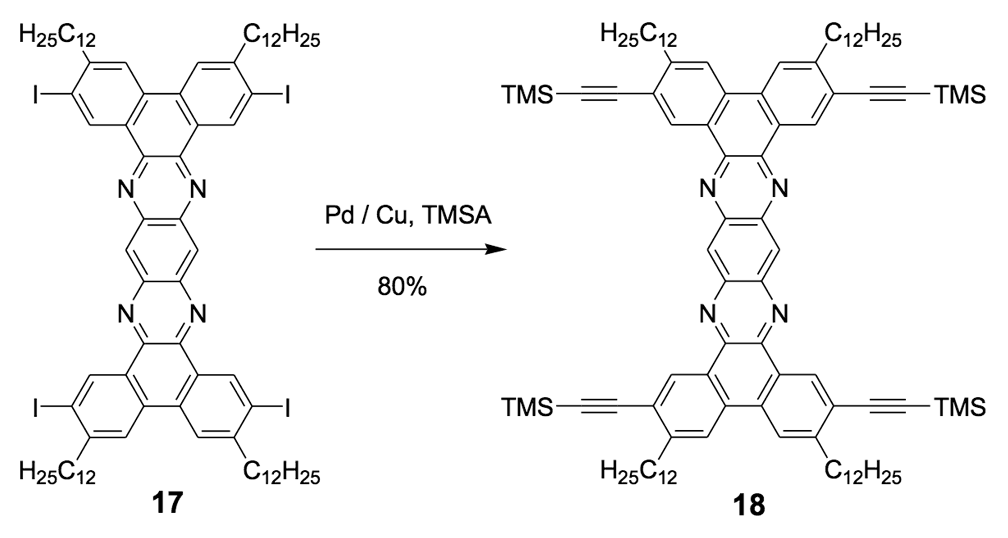
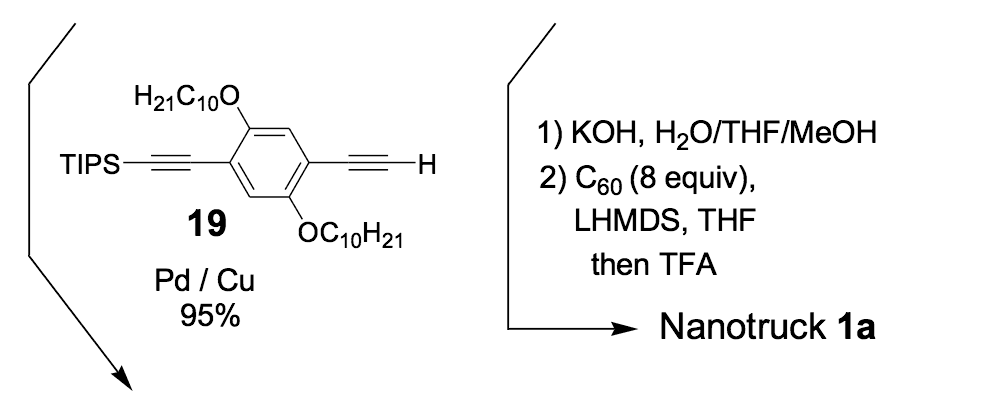
The use of excess LHMDS and C60 in THF is essential for the reaction. Nanotruck 1a remained insoluble in all organic solvents, which prevented us from determining the yield of a pure sample.
The complete purification of nanotruck 1a was hampered due to its low solubility in most good organic solvents for fullerene derivatives, most obviously, CS2 and o-dichlorobenzene. Since the fullerene-attached products are the only low solubility components in the reaction mixture, washing the resultant solid from the mixture produced the brownish solid of nanotruck 1a. Although it was impossible to directly determine the purity of nanotruck 1a by solution phase NMR, or other solution phase techniques, solid state 13C NMR and MALDI-TOF MS (matrix-assisted laser desorption/ionization time-of-flight mass spectrometer) analysis can be used roughly to estimate purity. The formation of nanotruck 1a was first confirmed by its MALDI-TOF MS spectrum (Figure 4).
Figure 4.
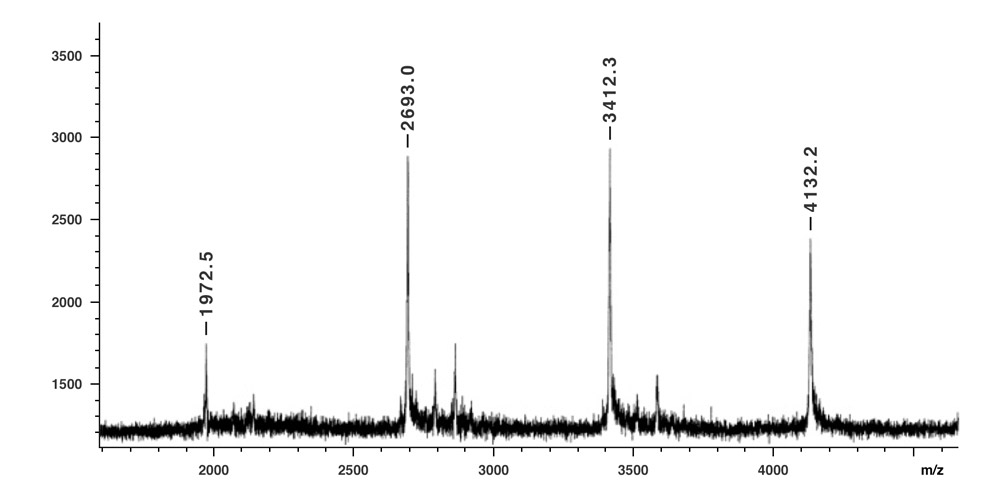
MALDI-TOF mass spectrum of nanotruck 1a. Peaks for one-, two-, three-, and four-wheel nanotrucks (m/z 1972.5, 2693.0, 3412.3, and 4132.2, respectively) are observed (negative ion mode, dithranol matrix).11
The molecular ion peak of nanotruck 1a is clearly observed at 4132.2m/z, while the signals at 3412.3, 2693.0, and 1972.5m/z are assigned to the fragmentations incurred by losing one, two, and three fullerene moieties, respectively, since the peak separations correspond to the mass of C60 (720Da) (Figure 4). In the solid state, 13C NMR experiments (and aside from the remarkably intense signals for fullerene and chassis aromatic sp2 carbons at ca. 150-120ppm, and the n-dodecyl carbons at 30.6, 23.6 and 14.4ppm), one can observe distinguishable resonance peaks for the sp3 carbons on the fullerene core.
The fullerene C-H carbon was assigned to the resonance at δ62.1 for several reasons. The signal intensity is significantly increased in the 1H-13C CPMAS experiment; the signal disappears in a dipolar dephasing experiment; the chemical shift is consistent with literature data. The quaternary fullerene carbon was assigned to the resonance at δ55.8 for this reason.
In Figure 5a, two alkynyl sp carbons are also discernable at δ83.1 (C60H-C≡C-) and δ97.5 (C60H-C≡C-), respectively, consistent with the solution state 13C NMR data obtained from other multi-fullerene structures.
Figure 5.
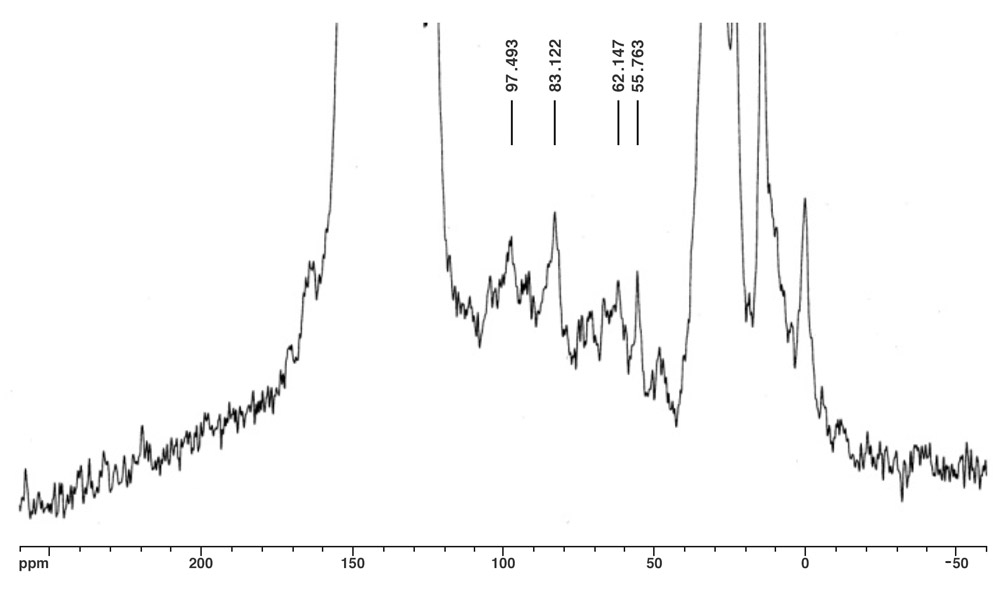
Figure 5a.
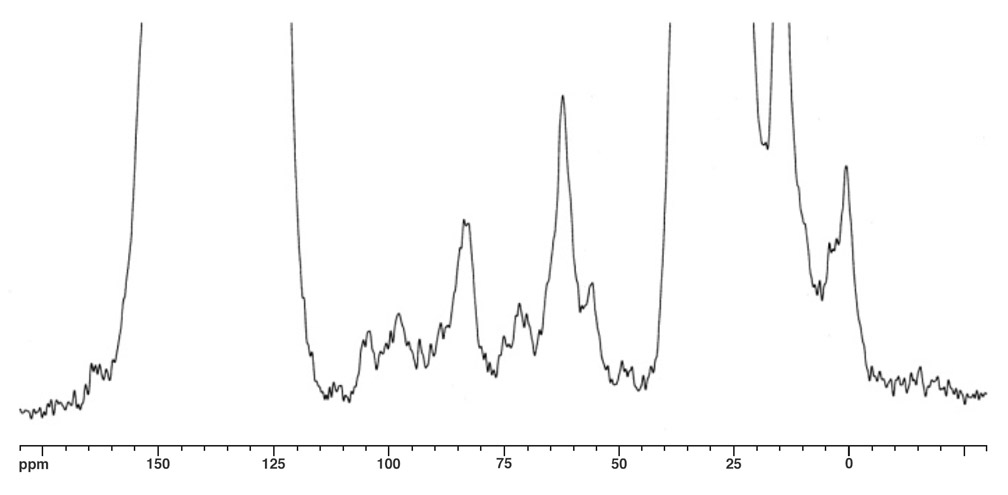
Figure 5b.

Figure 5c.
Solid state 13C and 15N NMR spectra of nanotruck 1a.12
Figure 5a. Direct 13C pulse, MAS at 6.8kHz.
Figure 5b. 1H-13C CPMAS, MAS at 6.0kHz, 3ms contact time.
Figure 5c. 1H-15N CPMAS, MAS at 4.0kHz, 10ms contact time, relative to glycine defined as ‑347.58ppm relative to CH3NO2 at 0ppm.
The proton attached to the fullerene is known to be significantly acidic due to its stabilized aromatic anion structure after deprotonation, and the chassis contains four nitrogen sites that could be protonated. 15N NMR can clearly differentiate between an imine and an iminium ion (protonation results in a large upfield shift); and for this reason, we carried out further experiments.
1H-15N CPMAS NMR experiments were conducted on nanotruck 1a and a model compound dibenzo[a,c]phenazine that possesses a similar nitrogen moiety. Nanotruck 1a shows a signal at –67.5ppm, which is consistent with that of the model compound at –66.7ppm. Analysis indicated that in the solid state, the fullerenes in nanotruck 1a are neutral rather than deprotonated.
The 15N spectrum could not be improved. Even with a full 7mm rotor, it took a week of signal averaging (118,000 scans with a 5s relaxation delay) to acquire this spectrum; with a molecular formula of C330H114N4, nanotruck 1a contains only 1.4 wt % nitrogen, corresponding to just 0.005wt % nitrogen-15.
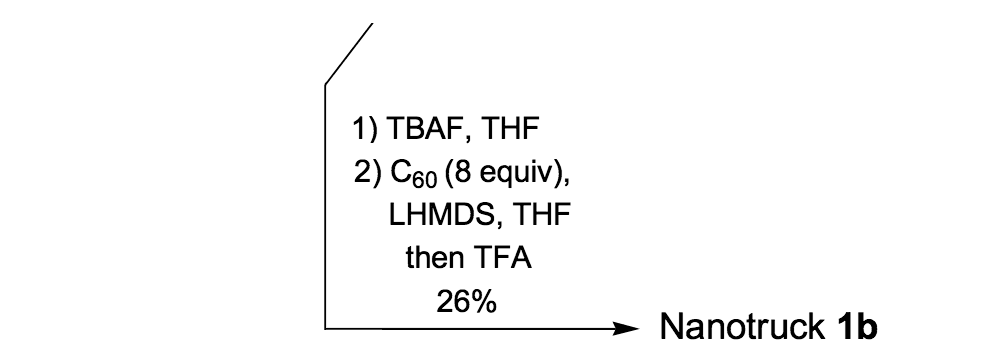
Because the four n-dodecyl functionalities failed to provide sufficient solubility for nanotruck 1a, we next tried to alter the axle structure by incorporating compound 19 (Scheme 2) to enhance solubility. The tetra-iodo full chassis 17 (Scheme 2) was coupled with compound 19, and then fullerene wheels were attached following the same in situ ethynylation procedure to yield nanotruck 1b with its better solubility.
Nanotruck 1b, it is satisfying to observe, could be properly purified and fully characterized.
The Synthesis of Nanocars
The tri-fullerene compounds 4a and 4b (Figure 1f and 1g) with OPE structure are relatively stable; soluble in common organic solvents; and they can be purified so that they are free of C60.
All to the good.
We thus combined our fullerene-wheel design with a Z-shaped OPE backbone to produce nanocars 3a and 3b (Figures 1d and 1e). Commercially available compound 21 (Scheme 3) was iodinated and coupled with TMSA using Pd-catalyzed conditions, and then the amino group was converted to an iodide to yield compound 22. The subsequent coupling reaction with axle unit 19 gave bromide 23. After replacing the bromide with iodide using a lithium-halogen exchange reaction, compound 24 was coupled with the middle chassis 25 to yield compound 26. The exchange reaction of the bromide group with iodide was required for a successful coupling reaction with 25.
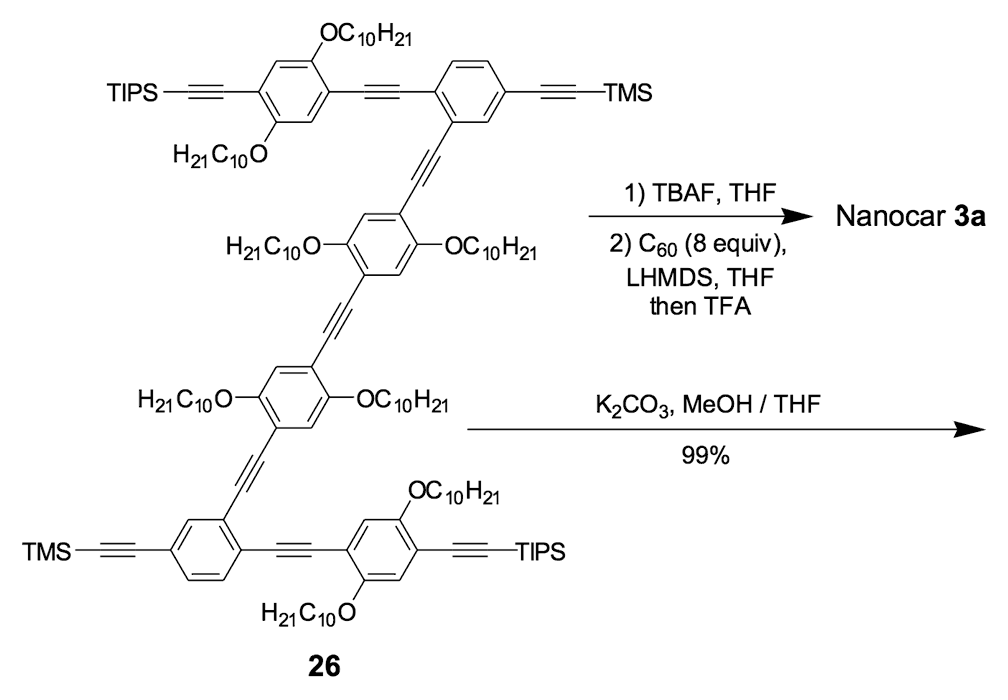
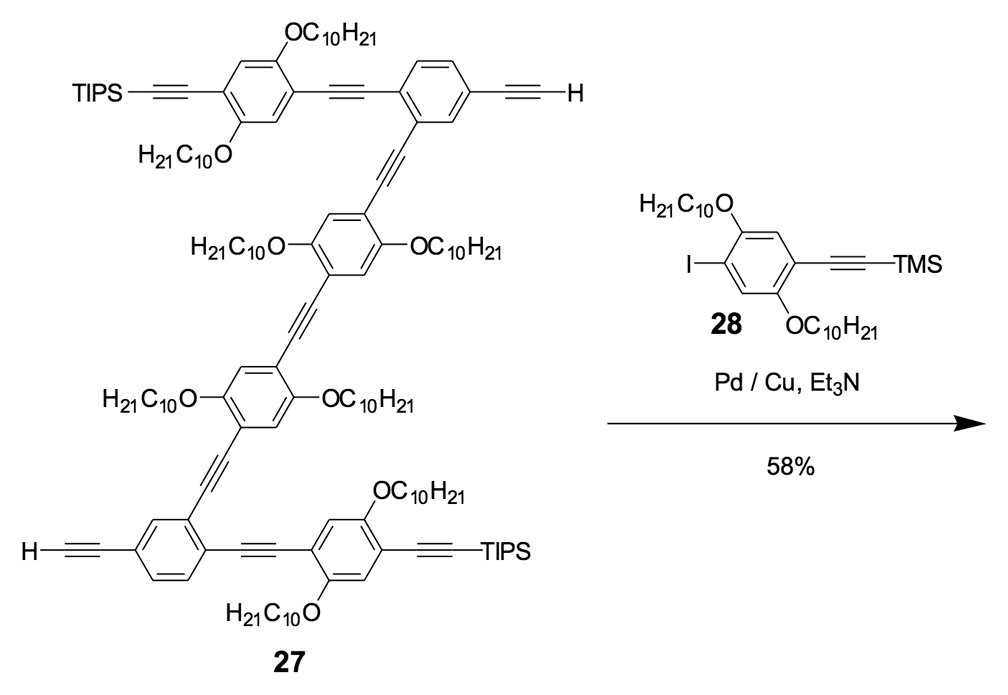

For all that, our attempt to get a pure nanocar 3a failed due to the limited solubility of the product. A crude, brown, solid material was obtained after the removal of solvents, but it was not soluble enough for further purification and solution-phase characterization.
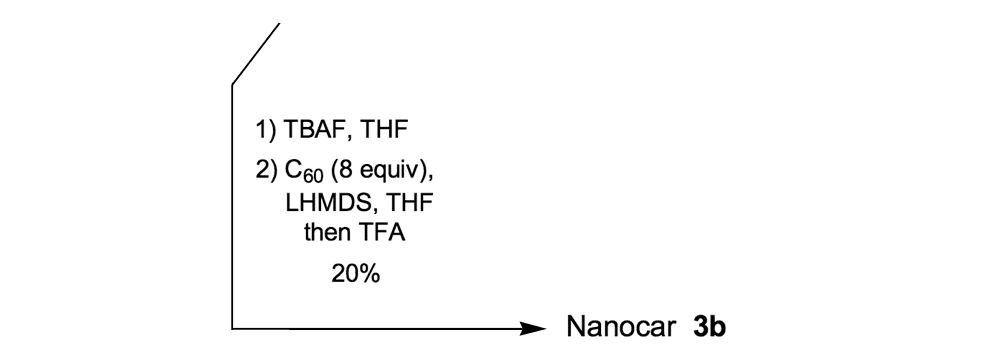
To further improve the solubility of our nanocar, another solubility enhancing group 28 (Scheme 3) was coupled after chemoselectively removing the TMS protecting groups using K2CO3. The coupling reaction with compound 28 yielded the full chassis 29. After the removal of all silyl-protecting groups with TBAF, four fullerene wheels were successfully coupled via the in situ ethynylation method to complete the synthesis of nanocar 3b.
Nanocar 3b was an improvement over nanotrucks 1a and 1b in terms of stability, solubility, and purity, and it was well suited for STM studies.
A triumph. At last.
Fullerene Motion
With nanocar 3b and the three-wheelers 4a and 4b, we have been able to demonstrate the action of fullerene-wheel architecture at the single-molecular level. Evidence for wheel-assisted rolling motion in the nanocar 3b on gold was obtained by comparing two different modes of thermally induced motions: translation and pivotal rotation (Figure 6).
Figure 6.
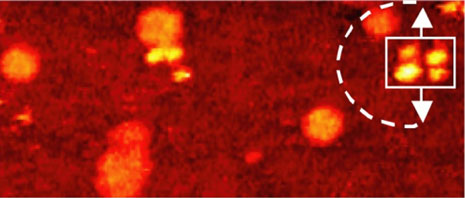
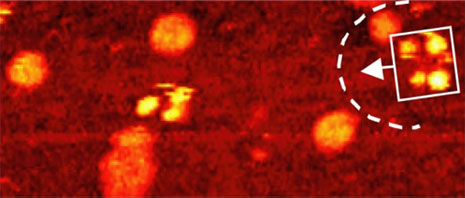
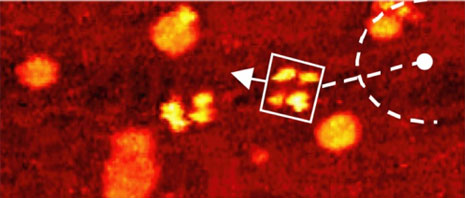


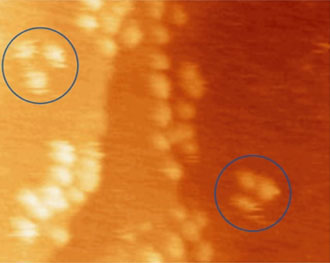



Comparison of thermally induced motions of four-wheeled 3b (Figure 6a) and its STM-imaged motions (Figures 6b–f), and three-wheeled 4a (Figure 6g) and its STM-imaged motions (Figures 6h–k). All sequence images were taken during annealing at ~200°C (bias voltage Vb = ‑0.95V, tunneling current It = 200pA. Image size is 51 × 23nm).
The orientation of the nanocar 3b is easily determined by the fullerene wheel separation, with motion occurring perpendicular to the axles.
Acquisition time for each image is approximately 1 min, with Figures 6b–f selected from a series spanning 10min, which shows ~80° pivot (Figure 6b) followed by translation interrupted by small-angle pivot perturbations (Figures 6c–f).
Figures 6h–k. A sequence of STM images acquired approximately 1 min apart during annealing at ~225°C show the pivoting motion of nanocar 4a (both circled molecules) and lack of translation of any molecules. (Vb = ‑0.7V, It = 200pA. Image size is 34 × 27nm).14
The four-wheeled nanocar 3b remained relatively stationary up to approximately 170°C. As the temperature increased, the molecules began to move in two dimensions by translating and pivoting themselves, and not in the 1D manner initially expected. At approximately 200°C, nanocar 3b remained, on average, slow enough to be followed through a series of 1-min images. Pivoting can be seen in a sequence of images (Figures 6b–f). The translational motion that occurred between pivoting was perpendicular to the axles, illustrating a directional preference relative to the molecular orientation (the length differing from the width permitted the directional assignment).
Above approximately 225°C, the rapid and erratic motion of the molecules could not be tracked due to the relatively slow acquisition time necessary (approximately 1min for a 90 × 90nm scan) compared to the rate of surface diffusion of the molecules.
When the three-wheeled nanocars 4 (Figure 6h–6k) were slowly heated to 225°C, occasional surface diffusion was observed. Translation in these cases was on the order of only a few nanometers over 20–30min for those cars that were translated at all. The majority of these molecules pivoted in place around a central point (Figures 6h–k). No significant translation or pivoting motion of trimer 4b was observed due to the wheel orientation relative to the axles. The pivoting behavior of trimer 4b continued even up to 300°C, a temperature at which the four-wheeled nanocars 3b were moving across the surface too quickly to be imaged by STM.
One would expect the energy barrier for sliding or stick-slip motion for nanocars 4a and 4b to be comparable to that of nanocar 3b, given that there is one less fullerene in 4a and 4b and thus a weaker overall interaction between the molecule and the gold surface. Since nanocars 4a and 4b exhibited little of the thermally induced translation over the temperature range investigated, this suggests that the motion of nanocar 3b is due to its rolling fullerene wheels.
Descent into Detail
The work that I have just described is merely an overview. The details are far richer. Consider the protocols for the conversion of compound 29 (Scheme 3) to nanocar 3b and their partial characterization data.15
Protocol 1
General Methods. All reactions were performed under an atmosphere of nitrogen unless stated otherwise. Reagent-grade diethyl ether and tetrahydrofuran (THF) were distilled from sodium benzophenone ketyl. Triethylamine (TEA) was distilled over CaH2. Fullerene (99.5+% pure) was purchased from MTR Ltd. And used as received. LHMDS (1M solution in THF) and TBAF (1M solution in THF) were obtained from Aldrich. Flash column chromatography was performed using 230–400 mesh silica gel from EM Science. Thin layer chromatography was performed using glass plates pre-coated with silica gel 40 F254 purchased from EM Science. Solution state 1H and 13C NMR spectra were recorded on 400 and 500 MHz spectrometers. Solid state NMR spectra were acquired at 200.13 MHz 1H, 50.33 MHz 13C, and 20.28 MHz 15N. Melting points were uncorrected. Ultra-sonicated fullerene slurry in THF was prepared in general ultrasonic cleaners.
General Procedure for the Addition of C60 to Terminal Alkynes Using LHMDS, in situ ethynylation method. To an oven-dried round bottom flask equipped with a magnetic stir bar was added the terminal alkyne and C60 (2 equiv per terminal alkyne H). After adding THF, the mixture was sonicated for at least 3 h. To the greenish-brown suspension formed after the sonication was added LHMDS dropwise at room temperature over 0.5 to 1.5h. As the reaction progressed, the mixture turned into a deep greenish-black solution. During the addition of the LHMDS, small aliquots from the reaction were extracted and quenched with TFA, dried, and re-dissolved in CS2 for TLC analysis (developed in a mixture of CS2, CH2Cl2 and hexanes). Completion of the reaction was confirmed by the disappearance of the starting materials. The reaction usually completed within 1.5 h from the beginning of LHMDS addition. Upon completion, the reaction was quenched with TFA to give a brownish slurry. Excess TFA and solvent were then removed in vacuo to afford a crude product that was purified by flash column chromatography (silica gel). Eluents and other slight modifications are described below for each compound.
Nanocar 3b. To a solution of compound 29 (Scheme 3), (0.096g, 0.030mmol) in THF (5mL) was added dropwise TBAF (0.2mL, 0.2mmol). 10 min after the addition of the TBAF, the reaction was quenched with saturated aqueous NH4Cl, and extracted twice with hexanes. The organic portion was dried over MgSO4 and filtered. After concentration in vacuo, the residue was purified by flash column chromatography with 30–42% CH2Cl2 in hexanes to give desilylated product (0.072g) as a yellow oil. This material was pure enough to carry on to the next reaction. The desilylated product (0.070g, 0.025mmol) was subjected to the general in situ ethynylation procedure with C60 (0.15g, 0.21mmol), THF (100mL), LHMDS (0.7mL, 0.7mmol), and TFA (0.7mL). (Note: product spot was not clearly visible on TLC analysis.) Crude products were dissolved in CS2 and directly loaded onto a column. The column was eluted with CS2/CH2Cl2 (100:1) to remove unreacted C60, and then with CS2/CH2Cl2 (1:1) for complete removal of trace C60 and elution of product. The product was further purified using another flash column with graduate elution of CS2/CH2Cl2/Hexanes (1:1:100), (3:2:5), then (3:3:4) to afford nanocar 3b (0.028g, 20%) as a brown solid. FTIR (CH2Cl2 cast) 2922, 2850, 2203, 1502, 1463, 1214cm-1; 1H NMR (500MHz, CDCl3) δ 7.77 (d, J = 1.4 Hz, 2H), 7.58 (d, J = 8.0Hz, 2H), 7.51 (dd, J = 8.0Hz, 1.4Hz, 2H), 7.30 (s, 2H), 7.25 (s, 2H), 7.18 (s, 2H), 7.15 (s, 2H), 7.14 (s, 2H), 7.10 (s, 2H), 7.07 (s, 2H), 7.03 (s, 2H), 4.19–4.14 (m, 8H), 4.09 (m, 4H), 4.00 (m, 4H), 3.90–3.88 (m, 8H), 1.95–1.13 (m, 192H), 0.88–0.80 (m, 36H); 13C NMR (125MHz, CDCl3) δ154.7, 154.6, 153.9, 153.75, 153.67, 153.65, (6 signals from aryloxy sp2-C in the aromatic ring), 151.65, 151.59, 151.497 (×2), 147.7 (×2), 147.44, 147.43, 146.74, 146.69, 146.50, 146.477 (×2), 146.46, 146.32 (×2), 146.31 (×2), 145.92, 145.89, 145.82, 145.77, 145.72, 145.69, 145.55, 145.529 (×2), 145.51, 145.45, 145.43, 144.78, 144.76, 144.60, 144.58, 143.28, 143.26, 142.69, 142.67, 142.66, 142.64, 142.21, 142.17, 142.13, 142.11, 142.07, 142.05, 141.98, 141.95, 141.77, 141.73, 141.69, 141.67, 140.47, 140.44, 140.40, 140.37, 136.18, 136.15, 135.3, 135.2, (30×2 signals from sp2-C in the C60 core), 134.4, 131.6, 130.9, 126.5, 125.8, 123.3, 117.6, 117.4 (×2), 117.2, 117.0, 116.9, 114.9, 114.7, 114.4, 114.2, 113.4, 113.2, 97.8, 96.1, 94.3, 94.1, 93.1, 92.2, 91.8, 91.0, 88.3, 80.2 (×2), 70.14, 70.10, 69.8, 69.6, 69.5, 69.3, 62.02, 61.98 (CH in the C60 core), 55.60, 55.59 (quaternary sp3-C in the C60 core), 32.04, 31.97, 31.95, 29.8, 29.74, 29.70, 29.66, 29.48, 29.45, 29.40, 22.77, 22.75, 22.73, 14.23, 14.21, 14.19; MALDI-TOF MS m/z (Sulfur as the matrix) calcd for C430H274O12 5632, found 5631 (M+).
These procedures are abbreviated and they are incomplete.16 A skilled synthetic chemist will know what the abbreviations mean and how to complete the uncompleted steps. A simple statement such as, “All reactions were performed under an atmosphere of nitrogen unless otherwise stated,” means that most of those reactions would have failed if done in the open air. Pre-treatment of solvents was needed so that the system would not be contaminated by impurities, such as oxygen, which retard or mitigate the desired reactions. Purification was required at each step since the chemistry rarely affords the chemist materials that are of sufficient purity for use in subsequent steps.
Each product needed a different purification protocol.
Chance may favor the prepared mind, but good things rarely happen only by chance.
How does nature do it? Are the biologists sure that they know the answer?
Are they very sure?
Cars with Motors
Our first-generation vehicles operated without motors.17 We later made motorized versions that were more complex to design and synthesize.18 The motors are ultraviolet-light-active; they rotate unidirectionally. The unidirectional rotation is derived from two elements of chirality: the atropisomerism of the double bond and the stereogenic center at the methyl site. Upon excitation, the double bond moves into an orthogonally arranged orientation, and the two directions of relaxation become diastereotopic. The molecules follow the lower energy direction. This is followed by a thermal helix inversion from a less to a more stable twisted alkene. This completes a half cycle. The process is repeated to complete a 360° rotation cycle.
The first motorized versions that we made (Figure 7e) rotated at 1.8 revolutions per hour. Too slow. But with a redesign, which took us back to step one in the synthesis, we achieved a light-activated nanocar whose motor spins at 3 MHz (3 million rotations per second) (Figure 7f).19 Fast enough.
Figure 7.
-
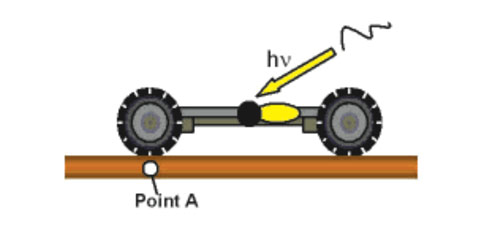
Figure 7a.
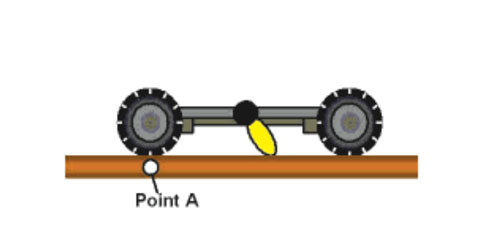
Figure 7b.

Figure 7c.
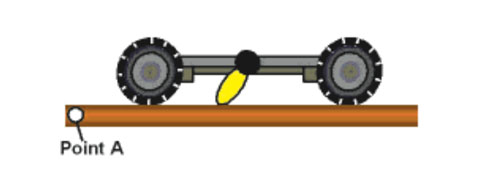
Figure 7d.
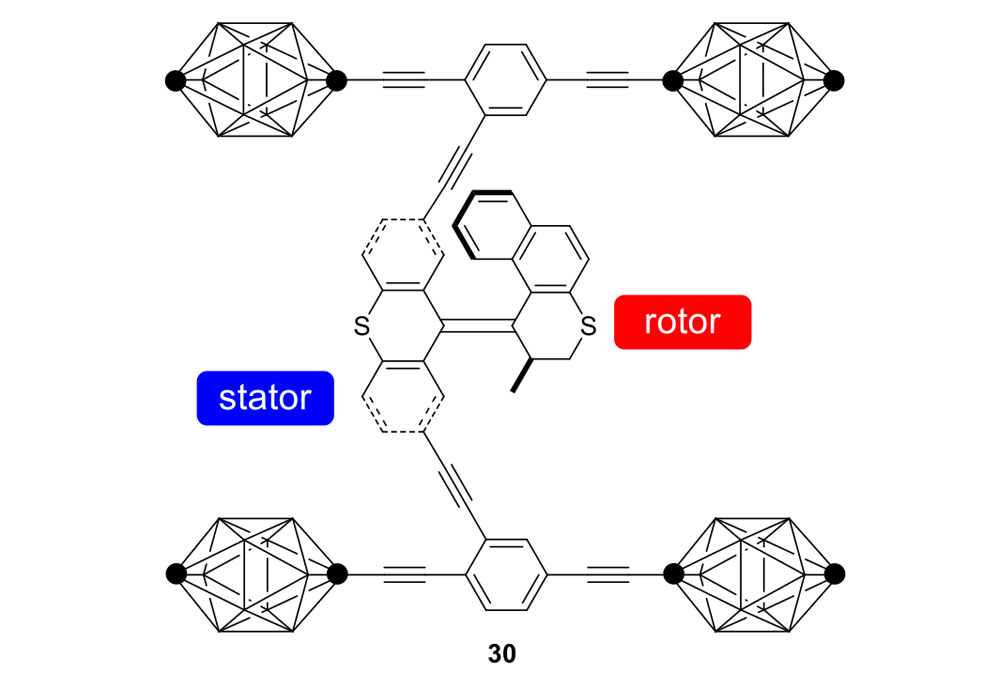
Figure 7e.
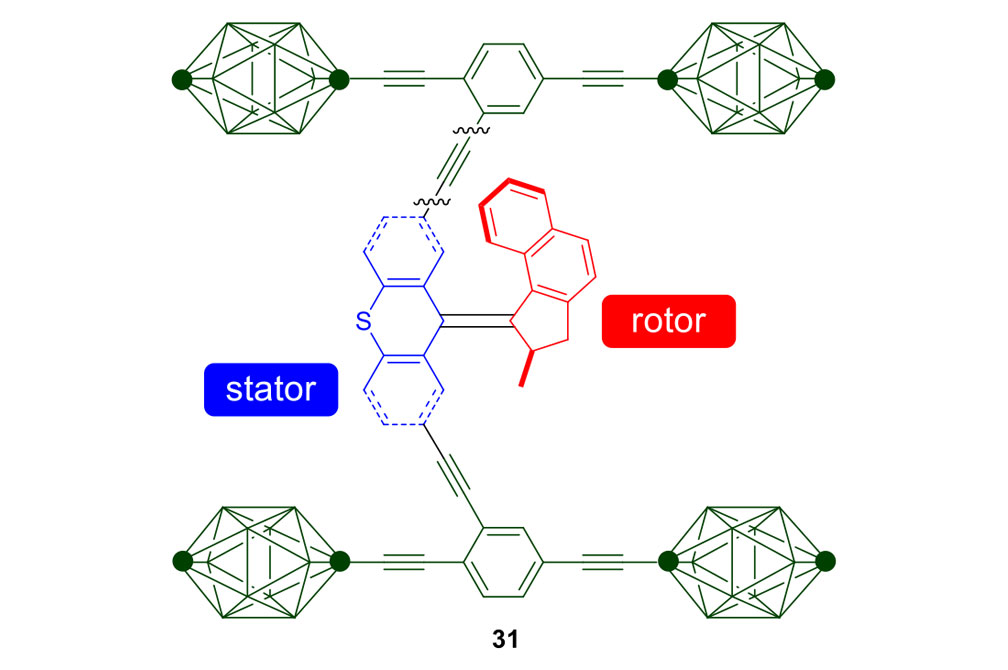
Figure 7f.
Figures 7a–d show the scheme of light-actuation of the motor so that it acts as a paddle-wheel to propel the nanocar along a surface.
The first-generation nanocar (Figure 7e) had a motor that only spins at 1.8 revolutions per hour while the second-generation nanocar (Figure 7f) motor spins at 3 million rotations per second. These nanocars have para-carborane wheels where they have BH at every intersection except the black pointed vertices, which represent C and CH positions, ipso and para, respectively, relative to the alkyne.20
Fast Motor Nanocars
Our plan to synthesize nanocar 31 (Figure 7f) involved a modular approach in which the coupling of the axles and the stator represented the last step. According to Scheme 4, heating ketone 32 to reflux in an ethanol and hydrazine solution produced the rotor, hydrazone 33. The conversion of ketone 34 into thione 35 was improved by decreasing both the concentration and the reaction time from those in the published procedure. The generation of the sterically hindered double bond between the rotor and the stator utilized Barton–Kellogg coupling. Hydrazone 33 was oxidized to the unstable diazo intermediate 36 using manganese dioxide by careful temperature control. The inorganic residue was removed by filtration in a Schlenk set-up that enforced the strict exclusion of air, oxygen and moisture. Thione 35 was added to the deep purple filtrate. A [2+3] cycloaddition occurred and evolution of nitrogen gas indicated the formation of episulfide 37. The white solid episulfide 37 was then treated with trimethyl phosphite in a screw-capped tube at 130°C to yield the molecular motor 38 as a mixture of isomers. We planned all along to investigate individual molecules on surfaces. It was unnecessary to resolve the enantiomers.
Nature would never have had this convenient luxury.


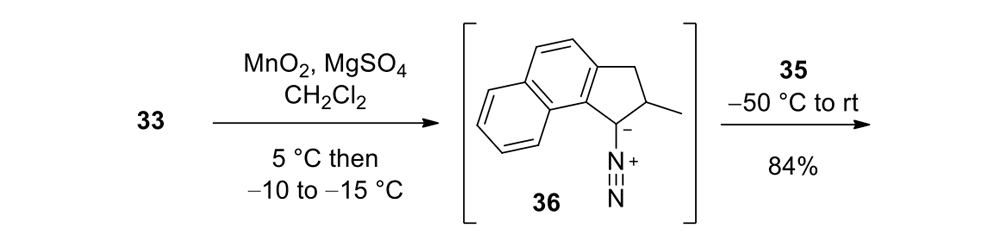
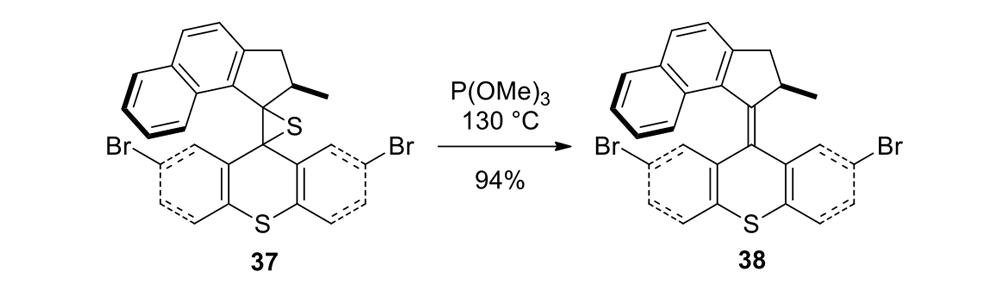
The synthetic details of episulfide 37 synthesis from compounds 35 and 36 are reproduced here so as to illustrate the precise temperature and atmosphere controls that are needed to assemble complex organic molecules.
Episulfide 37
To an oven-dried three-neck round-bottom flask charged with hydrazone 33 (0.99g, 4.7mmol) and MgSO4 (0.49g, 50% w/w) was added dichloromethane (25mL). To this suspension was added quickly MnO2 (1.62g, 18.8mmol, Sigma-Aldrich > 90%) at ca. 5°C. The reaction flask was immediately immersed and stirred in a cold bath ranging from −15°C to −10°C for 1.5h. After this period, the reaction mixture was cooled to −50 °C and then transferred to a Schlenk filtration tube connected to an oven-dried three-neck round-bottom flask. The deep purple filtrate that contained intermediate 36 was collected, and the Schlenk tube was rinsed with pre-cooled dichloromethane (20mL, −50°C). To the flask containing the combined filtrate, thione 35 (0.97g, 2.5mmol) was added portionwise until no more N2 evolved. The mixture was stirred for an additional 0.5h at ambient temperature. The mixture was poured into methanol (80mL) with vigorous stirring and a white precipitate formed. The solid was filtered, and the filter cake was washed with methanol (30mL) and dried under vacuum to afford the desired episulfide 37 (1.19g, 84%): mp 204°C (decomp); FTIR (neat) 3078, 3070, 3050, 2974, 2954, 2934, 2898, 2866, 2840, 1616, 1580, 1568, 1556, 1514, 1456, 1436, 1382, 1372, 1252, 1212, 1160, 1132, 1112, 1080, 1052, 1024cm−1; 1H NMR (400MHz, CDCl3) δ 8.89 (d, J = 8.8Hz, 1H), 8.00 (d, J = 2.0Hz, 1H), 7.83 (d, J = 2.0Hz, 1H), 7.56 (d, J = 8.2Hz, 1H), 7.54 (d, J = 7.8Hz, 1H), 7.40–7.34 (m, 2H), 7.29 (d, J = 8.2Hz, 1H), 7.26–7.20 (m, 1H), 7.18 (d, J = 8.2 Hz, 1H), 6.84 (dd, J1 = 8.2Hz, J2 = 2.0Hz, 1H), 6.77 (d, J = 8.2Hz, 1H), 3.43 (dd, J1 = 6.6Hz, J2 = 15.4Hz, 1H), 2.44 (d, J = 15.6Hz, 1H), 1.57 (qd, J1 = 6.9Hz, J2 = 6.6Hz), 1.12 (d, J = 6.9Hz, 3H); 13C NMR (100MHz, CDCl3) δ142.5, 140.9, 136.2, 135.3, 134.4, 134.1, 132.6, 131.8, 130.81, 130.76, 130.1, 129.7, 129.4, 128.2, 127.8, 127.6, 124.5, 124.4, 124.0, 123.4, 120.9, 120.0, 72.2, 60.8, 40.5, 38.1, 21.8. HRMS (APCI) m/z calcd for [M+H]+ C27H19Br2S2 564.9295, found 564.9275.
We confirmed the structural integrity of the key intermediate motor 38 (Scheme 4) using NMR techniques. All of the protons of motor 38 were unambiguously assigned with the assistance of correlation spectroscopy (COSY), nuclear Overhauser enhancement spectroscopy (NOESY), distortionless enhancement by polarization transfer spectroscopy (DEPT), heteronuclear single-quantum correlation spectroscopy (HSQC) and heteronuclear multiple-bond correlation spectroscopy (HMBC) experiments.
Nature uses a procedure far more complicated than anything illustrated here.
Figure 8.
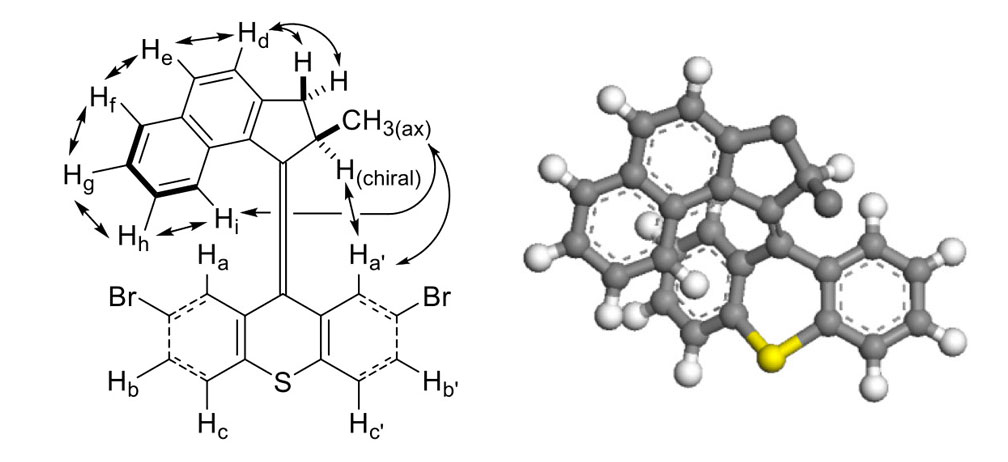
Figure 8a. (left) and Figure 8b. (right)
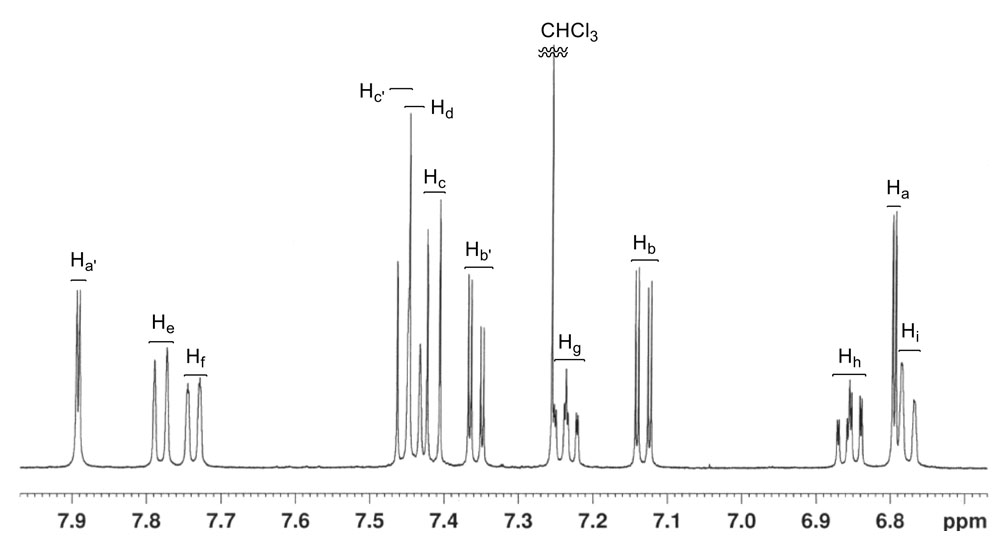
Figure 8c.
Figure 8a. Partial 1H NMR (500 MHz, CDCl3) at 295K of molecular motor 38. Selected NOE signals (double-headed arrows) were used to assign individual proton signals in the aromatic region as indicated in the top left structure.
Figure 8b. Ball-and-stick model of the energy-minimized (R)-isomer of motor 38 (without bromides) (Materials Studio with forcite force field).22
Determining Molecular Structure
Molecular structure determination is involved and difficult. The chemical shift difference steadily decreases between corresponding protons on the two benzene rings on the thioxanthenene stator: δ (Ha, Ha’) = 1.10ppm, δ (Hb, Hb’) = 0.23ppm and δ (Hc, Hc’) = 0.04ppm. This is consistent with the steadily increasing distance of these pairs from the rotor. The chemical shifts of protons Hh, Hi and Ha reflect the shielding effect caused by the presence of an aromatic ring directly above Ha or directly below Hh and Hi. Compared to Hh and Hi, the most shielded aromatic protons in precursors 32 and 33 (Scheme 4) are at δ7.49–7.52 and δ7.32–7.38; compared to Ha, the most shielded aromatic protons in precursors 34 and 35 are at δ7.43 and δ7.46. Interestingly, no NOE signals were found between Ha, Hi and Ha, Hh; this can be rationalized by an appeal to the helical structure of the rotor as seen in Figure 8b. Proton Hi exhibits the most complex line-shape among the aromatic protons. Not only does Hi couple to Hf, Hg, and Hh, but the COSY experiment also revealed coupling between Hi and each of the CH2 protons.
These brief remarks provide a shortened version of what is needed to determine molecular structure. Here is the full assignment information for that one molecule.
Full Assignment Information for One Molecule
A combination of standard 1D 1H, 13C, and DEPT-135 13C experiments and standard 2D 1H-1H COSY, 1H-1H NOE, 1H-13C HSQC, and 1H-13C HMBC experiments obtained on a 500MHz spectrometer yielded the assignments for molecule 38 discussed. The 1H spectrum immediately differentiated the two CH2 protons from the aliphatic CH proton by the distinctive large geminal coupling constant (15.6Hz) exhibited by the CH2 protons at δ3.652 and δ2.664. Only the downfield CH2 proton exhibited a detectable J coupling (6.2Hz) to the methine proton (δ4.186), which, exhibited a five-line pattern because of additional J coupling (6.9Hz) to the methyl protons (δ0.832). The 1H and COSY experiments revealed two groups of signals that each contained three-coupled aromatic protons (Ha, Hb, Hc and Ha’, Hb’, Hc’), two coupled protons (Hd and He), and four coupled protons (Hf, Hg, Hh, Hi) two of which (δ7.737 and δ6.776) were also coupled to a proton (δ7.781) in the adjacent Hd-He ring. Ha-Hb-Hc ring protons or Ha’-Hb’-Hc’ ring protons: δ6.794, d (J = 2.10Hz) of d (J = 0.35Hz); δ7.132, d (J = 8.30Hz) of d (J = 2.10Hz); δ7.414, d (J = 8.30Hz) of d (J = 0.35Hz). Ha’-Hb’-Hc’ ring protons or Ha-Hb-Hc ring protons: δ7.357, d (J = 8.25Hz) of d (J = 2.10Hz); δ7.455, d (J ≈ 8.3Hz) of d (J ≈ 0.2Hz); δ7.892, d (J = 2.05Hz) plus unresolved fine structure. Hd-He ring protons: δ7.440, d (J ≈ 8.2Hz, broad signals); δ7.781, d (J = 8.15Hz, broad signals). Hf-Hg-Hh-Hi ring protons: δ6.776, d (J = 8.55Hz, broad complex signals); δ6.855, d (J = 8.55Hz) of d (J = 6.70Hz) of d (J = 1.25Hz); δ7.236, d (J = 8.15Hz) of d (J = 6.70Hz) of d (J = 1.15Hz); δ7.737, d (J = 8.15Hz) of d (J = 1.25Hz) of d (J ≈ 0.6Hz) plus unresolved fine structure. In the Ha-Hb-Hc and Ha’-Hb’-Hc’ spin systems, the protons exhibiting only small J values (≤ 2.1Hz) were attributed to Ha and Ha’ because the couplings were over at least four bonds. Hb (Hb’) was tentatively differentiated from Hc (Hc’) on the assumption that 4JHH to Ha (Ha’) would be larger than 5JHH to Ha (Ha’). The upfield CH2 proton exhibited correlations to Hd and He, while the downfield CH2 proton also exhibited correlations to two of the four protons (δ7.236 and δ6.776) in the Hf-Hg-Hh-Hi ring. The long-range couplings to both of the methylene protons presumably account for the breadth of the Hd and He signals. The complexity of the δ7.737 and δ6.776 signals (more than just a d of d of d from couplings within the Hf-Hg-Hh-Hi ring) arises from the long-range coupling of the δ7.737 and δ6.776 protons to the δ7.781 proton in the Hd-He ring and the long-range coupling of the δ6.776 proton with both of the methylene protons. The aliphatic methine proton (δ4.186) exhibited a correlation to the aromatic signal at δ7.892 but not to the signal at δ6.794. The NOE experiment revealed critical information on the spatial proximity of various protons. In particular: (a) the presence of an NOE between the proton at δ7.892 and various aliphatic protons, and the absence of an NOE between the proton at δ6.794 and various aliphatic protons, enabled secure assignments for Ha’ (δ7.892) and Ha (δ6.794) to be made. NOEs were observed between Ha’ and the aliphatic methine proton (δ4.186), between Ha’ and the methyl protons (δ0.832), and between Ha’ and the methylene proton at δ2.664. (b) The presence of an NOE between the aromatic proton at δ7.440 and each of the methylene protons and the absence of an NOE between the aromatic proton at δ7.781 and each of the methylene protons enabled secure assignments for Hd (δ7.440) and He (δ7.781) to be made. (c) The presence of an NOE between the aromatic protons at δ7.781 and δ7.737 enabled the latter signal to be assigned to Hf, which provided a secure starting point for sequentially assigning the signals of the Hg (δ7.236), Hh (δ6.855), and Hi (δ6.776) protons through NOE. Hi also exhibited an NOE to the methyl protons. (d) The presence of an NOE between δ6.794 and δ7.132 and between δ7.132 and δ7.414 confirmed the Ha (δ6.794), Hb (δ7.132), and Hc (δ7.414) assignments. Similarly, the presence of an NOE between δ7.892 and δ7.357 and between δ7.357 and δ7.455 confirmed the Ha’ (δ7.892), Hb’ (δ7.357), and Hc’ (δ7.455) assignments. (e) Among the aliphatic protons, the methyl protons exhibited an NOE to the methine proton and to each of the methylene protons; the methylene protons exhibited an NOE between themselves; but only one methylene proton (3.652) exhibited an NOE to the methine proton. This established their cis orientation and the cis orientation of the methylene proton at δ2.664 and the methyl group.
The DEPT-135 13C experiment immediately differentiated the CH and CH2 carbons and differentiated the aromatic CH carbons from the fully substituted aromatic and alkene carbons. The two closest aromatic CH signals differed by only 0.034ppm. The 1H-13C HSQC experiment then gave specific, pairwise 1H/13C assignments for the 12 aromatic CH, aliphatic CH, CH2, and CH3 groups (Ha δ6.794 / Ca δ131.542, Hb δ7.132 / Cb δ129.190, Hc δ7.414 / Cc δ128.664, Ha’ δ7.892 / Ca’ δ130.654, Hb’ δ7.357 / Cb’ δ129.109, Hc’ δ7.455 / Cc’ δ129.143, Hd δ7.440 / Cd δ123.738, He δ7.781 / Ce δ130.895, Hf δ7.737 / Cf δ128.062, Hg δ7.236 / Cg δ124.510, Hh δ6.855 / Ch δ125.003, Hi δ6.776 / Ci δ125.709, methine H δ4.186 / C δ37.763, methylene H δ3.652 and δ2.664 / C δ39.678, and methyl H δ0.832 / C δ19.642). The 1H-13C HMBC experiment provided at least partial assignments for the ten fully substituted aromatic carbons and the two fully substituted alkene carbons. The quaternary 13C signal at δ125.818 exhibited a distinctive set of correlations to Ha, Ha’, Hc, Hc’ that enabled it to be assigned to the alkene carbon in the sulfur-containing ring. Each of the three quaternary aromatic carbons in the Ha-Hb-Hc ring exhibited at least one correlation to Ha, Hb, or Hc (δ141.564 with Hc at δ7.414, δ134.322 with Hb at δ7.132 and with Ha at δ6.794, and δ120.611 with all three of these protons) and exhibited no other long-range correlations. Each of the three quaternary aromatic carbons in the Ha’-Hb’-Hc’ ring exhibited the corresponding correlations to Ha’, Hb’, or Hc’ (δ139.425 with Hc’ at δ7.455, δ134.785 with Hb’ at δ7.357 and with Ha’ at δ7.892, and δ120.354 with all three of these protons) and exhibited no other long-range correlations. The aliphatic methine carbon exhibited a correlation to Ha’ at δ7.892 but not to Ha at δ6.794 (just as the aliphatic methine proton exhibited a correlation in the COSY experiment to Ha’ but not to Ha). Only the signal at δ148.160 exhibited a long-range correlation to the methyl protons, which was the basis for assigning this signal to the alkene carbon in the five-membered ring. This carbon also exhibited correlations to the aliphatic methine proton and both methylene protons. Each of the quaternary carbon signals at δ134.084 and δ146.309 exhibited correlations to the aliphatic methine proton, both methylene protons, Hd, and He; these carbon signals are assigned to the two quaternary aromatic carbons in the five-membered ring. The signal at δ134.084 was tentatively assigned to the carbon next to the alkene because it, unlike the signal at δ146.309, exhibited long-range correlations to the Hf-Hg-Hh-Hi ring (4JCH to Hf at δ7.737 and 3JCH to Hi at δ6.776). Each of the aromatic CH signals at δ123.738 and δ130.895 exhibited long-range correlations to each of the methylene protons, consistent with their assignments above. The two remaining quaternary signals at δ128.597 and δ133.121 were assigned to the ring junction carbons in the naphthalene moiety. Each exhibited long range correlations to each of the six protons in the naphthalene moiety. The signal at δ128.597 was tentatively assigned to the ring junction nearest the five-membered ring because this carbon exhibited correlations to both methylene protons and a weak correlation to the aliphatic methine proton, while the signal at δ133.121 exhibited only a weak correlation to one of the methylene protons.
In nature, molecular structure is confirmed by using complex molecules, the process akin to a glove fitting a hand. Whence that glove? It, too, had to be derived from a biological synthesis, with confirmation of its structure by a yet another glove that recognized its precise sequence, shape, and stereochemistry.
Designing nanoncars is child’s play in comparison to the complexity involved in the synthesis of proteins, enzymes, DNA, RNA, and polysaccharides, let alone their assembly into complex functional macroscopic systems. There are apparently a great many gloves in nature.
Final Assembly
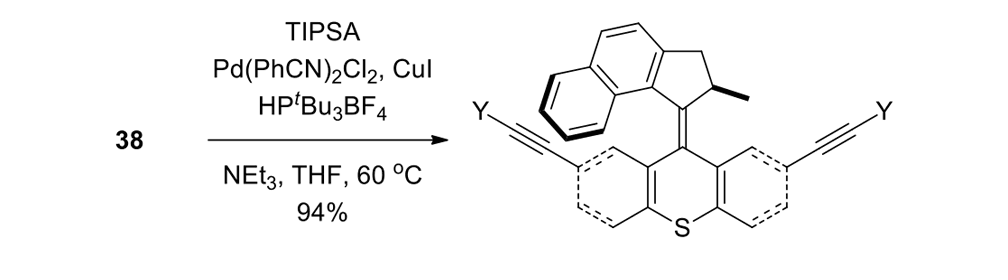
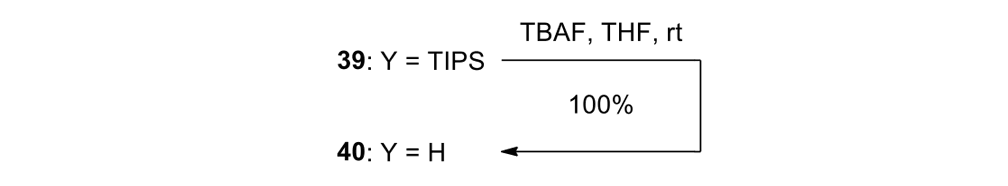
We attempted the final assembly of nanocar 31 (Scheme 5) using a method similar to our synthesis of the slow motor nanocar 30.23 The subtle structural differences between motors produced a remarkable difference in reactivities. Sonogashira coupling between TMSA and motor 38 using conventional Pd(PPh3)2Cl2 and CuI conditions did not afford the desired bis-coupled product. This was probably due to steric hindrance between the bromine atoms and the naphthalene rotor unit. A more reactive catalyst developed by Gregory Fu and his coworkers was used, but the result was disappointing because of a high degree of decomposition. Therefore, the more stable alkyne source, (triisopropylsilyl)acetylene (TIPSA) was mixed with dibromide 38. In a mixture of 6:1 NEt3/THF as base and solvent, Sonogashira coupling smoothly gave TIPS-protected bis-acetylene motor 39. The yield was excellent. Compound 39 had its TIPS groups removed using TBAF, producing dialkyne 40 in quantitative yield. Sonogashira coupling was used between dialkyne 40 and previously synthesized axle 41 using Pd(PPh3)4 and CuI as catalysts to produced nanocar 31 in moderate yield. Next, we tried a more convergent synthetic pathway (Scheme 5, Route II) utilizing Sonogashira coupling between motor 38 and alkynylated axle 42, and applying conditions analogous to those for the synthesis of motor 39.
Scheme 5.
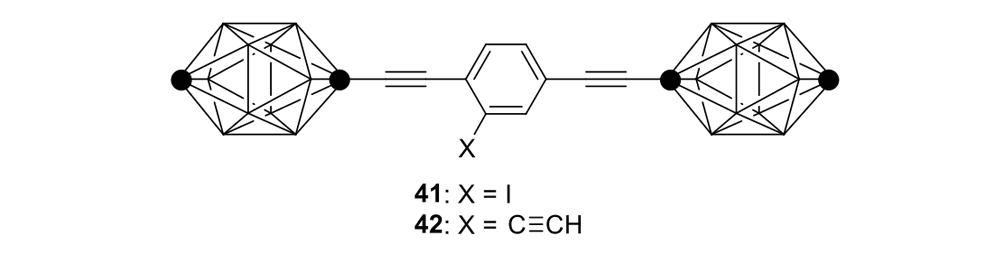
Synthesis of the second-generation motorized nanocar 31 through two different approaches.24
Motorized nanocar 31 was thus obtained but in lower overall yield than that obtained from Route I.
This underscores a common occurrence in organic synthesis. Even with modular approaches, small changes in the structure of the reactants make for enormous differences in reactivity. There is no simple work-around.
Slow to Fast
Consider the differences between motorized nanocars 30 and 31 (Figure 7). A small change in the rotors had an enormous impact on the rate of their unidirectional rotation, 1.8 revolutions per hour for nanocar 30 and three million rotations per second for nanocar 31. The rotor portion in nanocar 30 has a 6-membered ring bearing a sulfur atom, and the fast motor in nanocar 31 has a 5-membered ring bearing all carbons. What was involved in going from nanocar 30 to nanocar 31?
We had help. The motor as a basic unit has been studied for years by the Ben Feringa research group in the Netherlands.25 We knew this because their work has been published; we could build on their motors. Still, we could not use their motors precisely. We needed motors that would be suitable for new coupling reactions. The Netherlands gave us a prototype, nothing more.
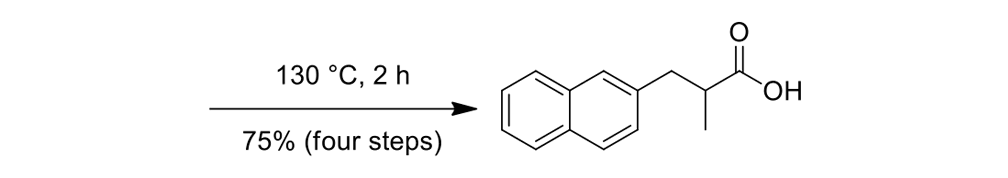
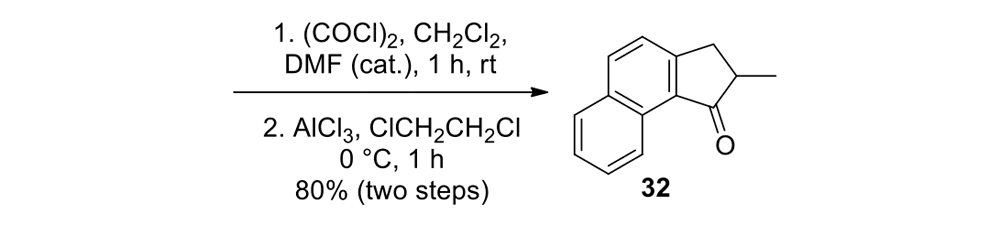
We thus had to start with 2,3-dihydro-2-methyl-1H-naphtho[2,1-b]thiopyran-1‑one (ketone 43, see Figure 9) for the slow motor and then use 2-methyl-2,3-dihydro-1H-cyclopenta[a]naphthalene (ketone 32, see Figure 9) for the fast motor.
But ketones 43 and 32 are derived from entirely different starting materials. There is no known method simply to expunge the sulfur atom in motor 43 and obtain ketone 32.
It is all very easy on the blackboard, of course; one can simply erase atoms at will and show ketone 43 becoming ketone 32 (Figure 9).
Figure 9.
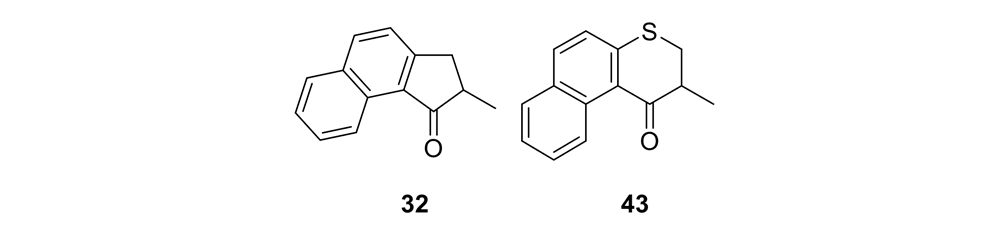
The starting ketones 32 and 43 for the synthesis of the fast motorized nanocar 31 and slow motorized nanocar 30, respectively. Removal of the sulfur atom in ketone 43 can, in theory, directly result in ketone 32. Though simple on paper using an eraser, there is no simple chemical methodology to effect that transformation.
As seen in Scheme 6, each had to be independently synthesized using entirely different starting materials and different routes.
Scheme 6.

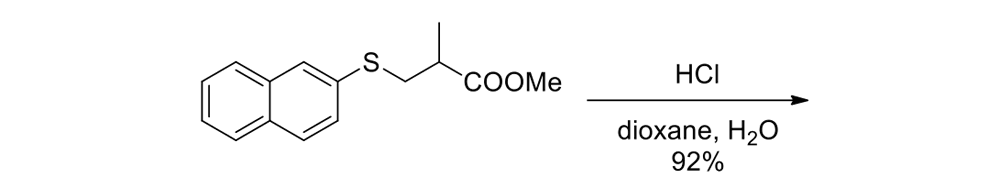
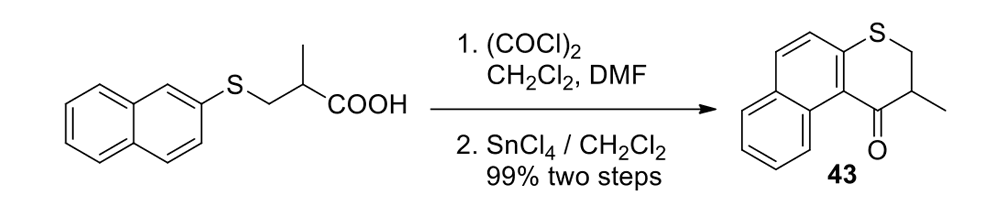
Scheme 6a.
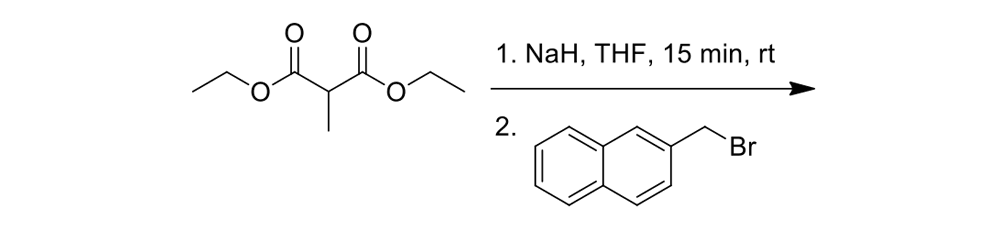
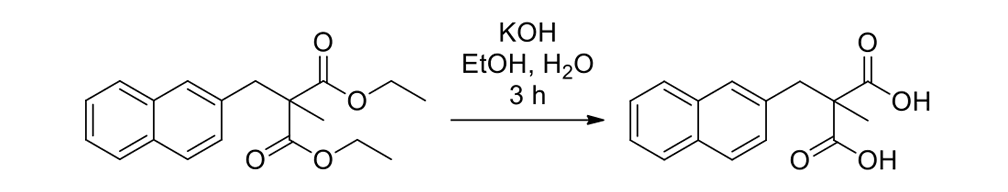
Scheme 6b.
Synthesis of the slow rotor (Scheme 6a) and the fast rotor (Scheme 6b) for the motorized nanocars.
Chemistry does not happen that way. Even the oxidative expulsion of SO2 from ketone 43 by the Ramberg–Bäcklund method would not work, since the sulfur is bound aromatically.
We could have left ketone 43 in a flask for millions of years and it would not form ketone 32 by any known or rational thermal, reductive, photochemical, or enzymatic method. This is not unusual when related compounds have clearly different starting points in organic chemistry. It is typical.
That nature has a select set of molecules that can be transformed into related structures using very precise enzymes (catalysts) is a phenomenon unknown in any other field of organic chemistry.
Wheel Changes
Why did we change the fullerenes wheels from nanocars in Figure 1 to the carborane-wheeled motorized nanocars in Figure 7? Because we had to. There is no way to achieve motor functionality in nanocars 30 and 31 using fullerene wheels. We did not know that until we built nanocar 30 with fullerene wheels. To our disappointment, when the motors are photo-excited, they immediately transfer their energy to the fullerene wheels so that the motors do not rotate.
We learned that after the fact.
Thus we had to change the wheels on the motorized cars to carboranes, since carboranes do not accept energy from the photo-excited motors because of the large energy difference between their lowest unoccupied molecular orbitals (LUMOs) relative to the excited state energy of the motor.
A key point: parts are not always easily interchangeable without severe and unexpected consequences.
A second key point: fullerene wheels are good for use on metal surfaces such as gold due to a strong charge transfer interaction between fullerenes and gold of ~45kcal/mol or ~200kJ/mol. This strong surface interaction causes the fullerenes to roll on the gold surface rather than to slide like a car on ice. But the carborane wheels, when used on metal surfaces, had very poor adhesion; the cars simply slid up against a one-atom-high ridge in the metal surface where a charging gradient was able to pull the nanocars. This can be exemplified in the nanodragster 44 (Figure 10a), where the methyl-tipped carborane wheels show so little adhesion to the surface that nanodragster 44 has its front carborane wheels lifted at a charged domain near a one-atom-high step edge on the gold surface.26
The darn thing popped a wheelie and flipped over to straddle the step edge (Figure 10b).
Figure 10.
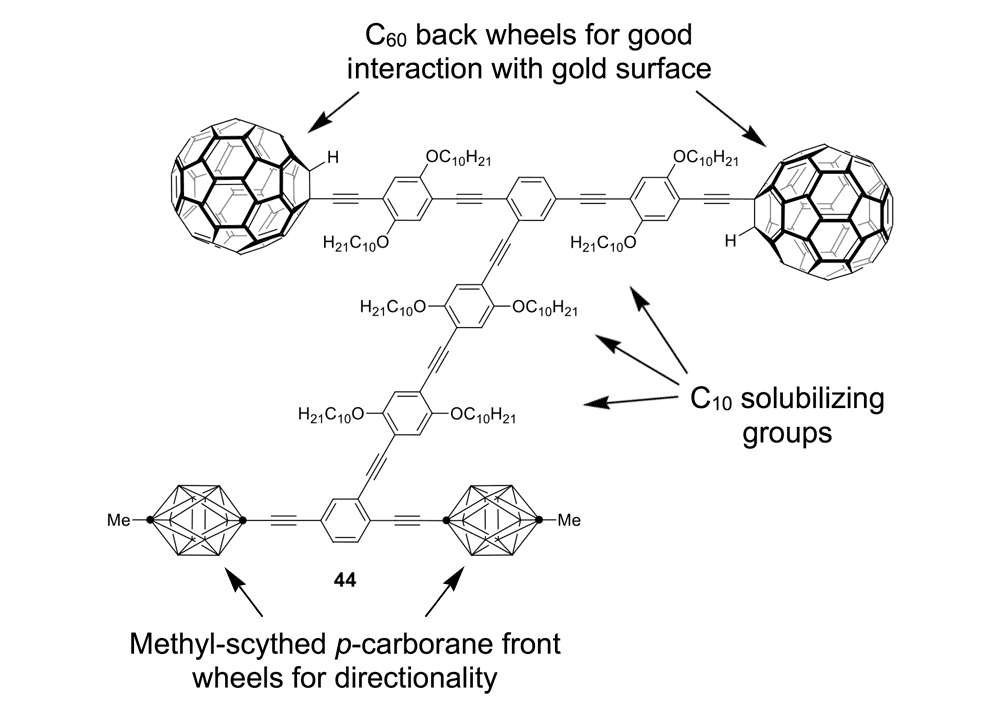
Figure 10a.

Figure 10b.
Figure 10a. Molecular structure of the nanodragster 44. The p-carboranes have BH at every intersection except at the points denoted by (·) which represents C positions.
Figure 10b. Left image: an STM micrograph of the of the nanodragster 44 where the two fullerene wheels are 3.3nm from center to center, and pinned along a one-atom-high step edge on gold. The brighter region is higher side and the darker region is the lower step edge. The carborane wheels, circled in black, rise up under the influence of the tip bias and “pop a wheelie” to flip over to the other side of the step edge as shown in the right image (carborane wheels circled in white). The horizontal axis of the pictures are 10nm.27
The carborane wheels were not suitable for use on metal due to their low adhesion. But the carborane wheels worked well on glass since there is a hydrogen-bonding interaction between the B-H units and the oxygen moieties on a glass surface; we have used them extensively to study nanocar motion on glass surfaces.28
When working environments change, drastic changes in molecular structure are often required to retain the system’s functions.
Checklist
- Determining a target?
Done. Molecular machines. - Insurmountable problems?
The first generation of nanovehicles had insurmountable solubility problems.
Although organic chemists have at least fifty widely differing solvents and solvent polarities from which to choose, we could find no satisfactory solution. - Starting over?
Yes, of course. - Redesign?
Done. Molecular flexibility, specifically a less rigid chassis, was needed (nanocars 1a and 1b to nanocars 3a and 3b) because restrictions at step edges prevented the nanovehicle from moving over long ranges.
When we added a motor to the nanocars, the former chassis proved insufficient. We had to redesign the intermediates in order to affix the motors. Luckily, we could store all new intermediates in the freezer to prevent their decomposition while we built the motors, and were able to access the published work of the Feringa group from the Netherlands in order to build upon their initial designs. But even then, starting at step one for the motors, we had to build in functional handles that would let them be accepted into the chassis and wheel assemblies. Only after constructing the new motors did we learn that they were incompatible with the well-developed fullerene wheels due to energy transfer problems. - Redo redesign?
When we desired to go from a slow motor to a fast motor, though the stator was reusable, the rotor was not. The rotor had to be redesigned, from step one, so as to become a faster unidirectional rotator. - And again?
Once we changed the wheels to make them suitable to work with the motors, we learned that the new wheels were not compatible with a gold surface. So we had to change to a glass surface. The glass surface was not compatible with our imaging technique by STM, which requires a conductive surface. Glass is an insulator. - Starting from scratch?
To image the nanocars on glass, we had to use a technique called single molecule fluorescence microscopy (SMFM), but another problem arose: the nanocars were not fluorescent. This meant appending a fluorophore to the chassis. But we first had to re-synthesize a new chassis to which the fluorophore appendage could be attached. We could have purchased a pre-synthesized fluorophore from a chemical company. These bulky fluorophores slowed the nanocars. So, starting from scratch again, we built a chassis with fluorescent axles (Figure 11). These we had to build using entirely different chemistry that we had used in the past cases. Sadly, motor energy transfer to the now fluorescent axles decreased the efficiency of the motors.
Newer versions have motor excitation frequencies optically far-separated from the fluorophore excitation frequencies. We know what frequencies to target because we can record the optical spectra using UV-visible absorbance and emission spectroscopies.
Figure 11.

Structure of a fluorescent (based on the boron-containing axles) and motorized nanocar. The nanocar is designed to move in circles due to the orientation of the axles with respect to each other. Notice that the wheel type changed here to adamantanyl groups which will further facilitate movement on glass based on our previous studies.
- Yes, but
Even our optimized yields were not quantitative (~100%). First time reaction yields were usually very low, sometimes as low as 0%. Only after repeated trials under different conditions could we attain a yield range of 50% to 60%, and sometimes not even that. - Well obviously
Most organic chemists would agree that even with extensive planning, 90% of reactions are failures. Substrates and conditions must be repeatedly modified to secure respectable and usable yields. At each step a massive amount of time is spent on separations and optimizations. If byproducts are permitted to accumulate, they can consume the new steps’ reagents and alter the course of the reaction. After every one or two steps there must be purification.
If all our reactions were near 100% yield, it would ease the separation problems. But this can take years to achieve, if it is possible at all. And even then, sufficient atom efficiency is very rare. Byproducts from the other reagents fill the system. High atom-efficient reactions are even harder to achieve. The loss of materials is expensive. In most cases, these byproducts cannot be converted back to usable compounds in an efficient way. Developing a scrubber system for degrading these products back to usable starting materials would, in most cases, take more time and money than developing the original target synthetic routes themselves. The separated byproducts are put into waste disposal containers and sent for destruction through combustion.
But we use petrochemicals as our major feedstocks, and these come in enormous amounts from fine-chemicals producers. Large amounts of energy come from power grids. - Don’t forget
We had the convenience of ordering many of the requisite reagents to start our syntheses. The just-in-time (JIT) procurement system permits us to have most chemicals at our doorstep within 18 to 24 hours. Even so, detailed planning and logistics go into making sure all reagents and solvents and gases and glassware are ready for a day’s lab work.
Solvents need to be pre-distilled before use since small impurities can promote or catalyze undesired side reactions. Intermediate molecules need to be pre-made and properly stored in a freezer away from light and oxygen to prevent their decomposition while the other segments of the synthesis are being done.
A rich chemical literature provides guidance on reaction types and conditions that are useable on similar molecular constructs, although modifications are almost always needed since the substrates in a new synthesis are different. - Now that I think about it
Reagent addition order is critical. A needs to be added before B, and then C, and each at its own specific temperature to effect a proper reaction and coupling yield. The parameters of temperature, pressure, solvent, light, pH, oxygen, moisture, have to be carefully controlled. Unless one can devise sophisticated promoters or catalysts that are stable in air and moisture and can work at common atmospheric conditions, precise control must be maintained. But making such ambient stable promoters or catalyst is more complex than just varying the temperature for the specific reagent, or putting the reaction in a carefully maintained atmospheric control box (dry-box) equipped with oxygen and moisture sensors, all maintained under a positive pressure of inert nitrogen gas. - It’s not cheap
Once the desired product is synthesized, it can take much longer to properly characterize the product than it did to make it. We use a host of tools, costing millions of dollars, to facilitate rapid molecular structure identification.
In chemistry, everything is hard, time-consuming, and expensive.
Life Lessons for the Prebiotic Chemist
Carbohydrates are the backbones of nucleotides, which in turn are needed for DNA and RNA. Carbohydrates also serve as recognition sites for cells to communicate with each other, and as food sources for living systems. The difficulties involved in carrying out carbohydrate synthesis in a prebiotic environment parallel those found in making nanovehicles.
Consider the pentose sugars (Figure 12). These sugars have three stereogenic centers, so there are eight possible isomers. Some substructures are enantiomers, others diastereomers; all are chiral.
Figure 12.
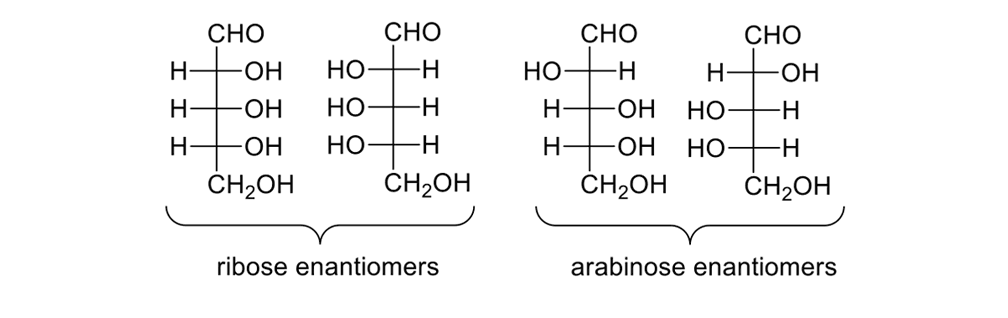
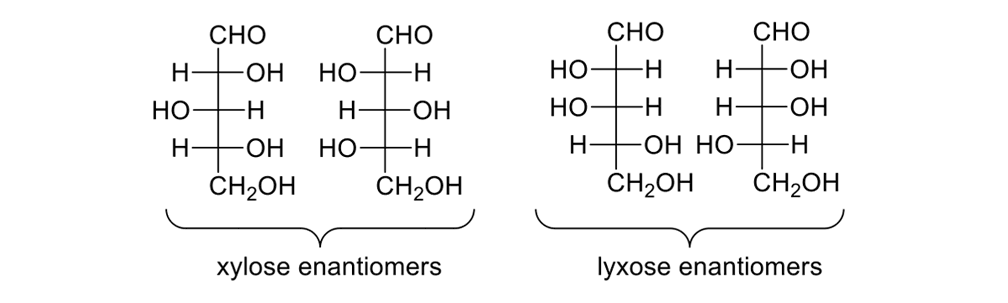
The eight pentose sugars showing the four sets of enantiomers depicted in Fischer projections.
DNA requires a five-carbon sugar, or D-(-)-ribose, one of eight possible pentoses. Blind synthetic pathways lead to a host of products that are unwanted because they are unneeded; and, yet, acting as blindly as Louis Braille, nature somehow found the requisite five-carbon sugar. How?
So far as life goes, as Teacher’s is the great Scotch, water is the great solvent. Nature depends on variations in pH and salinity. Organic synthesis is very hard to do in water. Highly oxygenated organic compounds are needed. The synthetic chemist must project the oxygenated groups out toward the water domain, and project the non-oxygenated groups in toward each other, thus generating a hydrophobic domain. It is very hard to do.
By doing our nanocar organic synthesis in organic solvents rather than in water, we markedly lessened the difficulty; it is a luxury that nature did not (and does not) enjoy. Starting from scratch, she would have had to redesign her structures, discarding the inevitable false starts as they occurred. Whatever else she may have been doing in the prebiotic era, nature was not consulting the modern chemical literature.
Any prebiotic system is destined, at least some of the time, to crash and burn. How does nature know how to stop, or why?
As for the yields of chemical reactions, we design the reactions to minimize diastereomeric mixtures that can be nearly impossible to separate. We try our best to avoid the undesired diastereomers because their separation is too time-consuming and expensive. They waste a huge amount of starting material; they generate unwanted products. Enantiomeric separations are all the more difficult. We avoided that by building a system with only one motor which functioned regardless of whether it turned clockwise or counterclockwise.
Nature has chosen a far harder route, using predominantly one enantiomer (homochiral) in a system with multiple stereogenic centers.
Tough for us, easy for her, strange all around.
Sugars and Eschenmoser
Albert Eschenmoser is a great synthetic chemist. He has spent years suggesting prebiotic routes to the five carbon pentoses. Direct synthesis, he discovered, (Figure 11) was not successful when starting with glycoaldehyde,29 and using an old-fashioned formose reaction in which a base is catalyzed with formaldehyde in the presence of a divalent cation such as calcium.30
Eschenmoser did not start with formaldehyde, nor with its dimer, glycolaldehyde, but with phosphorylated glycolaldehyde (glycoladehyde phosphate) (Figure 13).31
Figure 13.
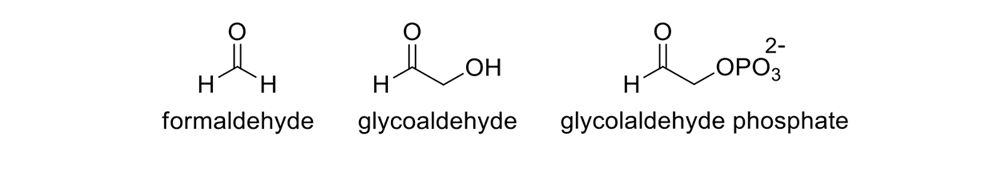
Three common starting materials in prebiotic chemistry research.
Since glycolaldehyde is the dimeric form of formaldehyde, he first had to make the dimer of formaldehyde. Only then could there be further aldol chemistry in the formose reaction. Nonetheless, a good organic chemist can design conditions that will isolate the product, purify it, and then proceed.
A very good organic chemist, this is what Eschenmoser did.32
Can phosphorylation then occur? Yes, but only given high concentration of a phosporylating agent. It does not happen in the presence of the strong base required for the formation of the glycoaldehyde itself.
The glycoaldehyde needs a strong base to pump up the reaction, and then, to stop it dead, it needs to be separated from that strong base.
While in a near-neutral aqueous solution, and in the presence of magnesium ions, the glycoaldehyde was phosphorlylated with four molar equivalents of amidotriphosphate. Once phosphorylation was complete, and the glycolaldehyde phosphate isolated and purified, Eschenmoser then exposed glycolaldehyde phosphate to the very basic, 2N sodium hydroxide at a concentration of 0.08 moles per liter.
The formaldehyde started life in a strong base; its product was isolated and freed from the strong base, and exposed to a neutral solution of amidotriphosphate (which the researchers made separately). That product, glycoladehyde phosphate, was then isolated and conveniently re-exposed to a strong base.
Even with this masterful design, the result was mostly undesired racemic hexose triphosphates. Using a stroke of superb synthetic insight, Eschenmoser placed the glycolaldehyde phosphate in the strong base, and then added 0.04 moles per liter of formaldehyde to obtain a 40-50% yield of mostly racemic pentose diphosphates (the mixture shown in Figure 13 but with two phosphates at C2 and C4 of each of the structures).
In that 40–50% yield mixture, there were then eight possible isomers (four diastereomers): the desired racemic ribose-2,4-diphosphate (~15% of the total from the reaction), racemic arabinose-2,4-diphosphate, racemic lycose-2,4-diphosphate and racemic xylose-2,4-diphosphate (where racemic signifies a 1:1 mixture of the two enantiomers) along with 11 other identified carbohydrate species, all bearing their enantiomer partners. That meant 22 other identified species from the 40–50%, with the remaining 50–60% being unidentified nonvolatile compounds such as higher oligomers and polymers.
Even Eschenmoser did not attempt to separate out the desired, albeit still racemic, ribose-2,4-diphosphate. And for very good reason: it would have been nearly impossible.
Biologists can easily imagine nature selecting the correct isomer. Not so synthetic chemists. Selected by what? No enzymes were yet available. The data more readily suggests that no prebiotic process is likely to yield the requisite carbohydrates.
The most masterful of synthetic chemists could produce only gross mixtures.
Eschenmoser remarks candidly that:
[I]f under otherwise standard conditions for the formation of the family of pentose-2,4-diphosphates one allows the reaction to run not simply for one week but rather for 23 weeks, the total amount of the four diastereomers [that is eight isomers] is largely unchanged, but the ratio of the diastereomers is displaced such that the content of ribose diphosphate is increasingly diminished in favor of arabinose diphosphate, with the latter ultimately gaining the upper hand. This observation accounts for the conclusion that the preferential formation of [the desired] ribose-2,4-diphosphate under the reaction conditions employed is a result of kinetic reaction control.33
This means that time works against life. Over a mere 23 weeks, the desired diastereomer—the racemic ribose-2,4-diphosphates—had been reduced from a 17% yield to a 7% yield. After a year, there would be very little left. In the laboratory, as anywhere else, it is essential to stop a reaction before the desired product degrades.
Of all the isomers, the one desired was not the most stable, even given the cleverly designed experiment that started with glycolaldehyde, and that was further intelligently and conveniently phosphorylated to the glycolaldehyde phosphate.
Racemic ribose-2,4-diphosphates degraded under the very controlled conditions under which it formed.
Eschenmoser writes that “the total amount of the four diastereomers is largely unchanged,” but his own data shows that the combined yield of the four diastereomers dropped from 34% to 30% over the course of the additional 22 weeks, a relative 12% loss of the pentoses over 22 weeks.34 After a few years, a brief moment in time in prebiotic terms, there would be few if any of the pentoses left, let alone the desired ribose-2,4-diphosphate.
Where does the material go? It likely degrades to extended oligomers and polymers, a process common in organic reactions, and especially common in extended aldol-based reactions.
After All That
His work on the pentoses ending in frustration, Eschenmoser switched to what he called homo-DNA, an oligonucleotide composed of d-2,3-dideoxyallopyranose. The work represented a demotion of his ambitions. “[F]rom an etiological perspective,” he wrote,
the work on homo-DNA was … a model study only, albeit one that proved to greatly facilitate our entry into base-pairing prognoses, the development of methodology for oligonucleotide preparation, and the interpretation of base pairing characteristics of alternative nucleic acids.35
If he could not get what he wanted, Eschenmoser determined to want what he could get:
Whenever possible, experiments on the etiology of potentially primordial biomolecular structures should be planned such that the results might not only have etiological relevance, but also class as valuable contributions to chemistry in general. Etiological relevance will ultimately remain uncertain; if sometime later it should succumb to new insights, the results still survive as a contribution to chemistry.36
Eschenmoser recognized the uncertainties inherent in prebiotic chemistry; he was looking to advance synthetic chemistry’s base of knowledge, an easier and more lasting contribution.
Wish Fulfillment
From a paper on prebiotic chemistry:
Moreover, there was the well-known—but still no less remarkable—fact that in cellular biochemical processes, monosaccharides apparently never operate in the free state, but always in phosphorylated form. It is a short step from such considerations to the notion of a primordial scenario in which, again, phosphorylated and not simply neutral forms of carbohydrates would have been operative [emphasis added]. In a self-organization process in a primordial environment, it may have been of primary importance for carbohydrate molecules to escape chemical chaos, finding themselves instead at concentrations suitable for chemical reactions, and in reaction spaces that would facilitate efficient chemical transformation. With respect to both requirements, phosphorylated sugar molecules would, through their electrical charges, have offered advantages over neutral, water soluble carbohydrates in environments containing mineral surfaces or minerals with expandable layer structures.37
That short step is not short at all. Biochemical routes are far downstream and occur in far more complex scenarios. In the laboratory, phosphorylation required precise control of phosphorylating agents.
These hopeful but unlikely suggestions pain the synthetic chemist under any circumstance, but for some remarkable reason, they are tolerated in prebiotic chemistry.
Despite claims to the contrary, research on mineral surfaces has done little to solve the problem of overall yields, or that of diastereo- and enantioselectivities.38 Reagent addition order is critical. An abiogenetic pathway would require several lines of intermediates forming in proximity, and then coming together in the proper order at the precise moment and location needed for synthesis. We could modify many parameters during synthesis: temperature, pressure, solvent type, light, pH, oxygen, moisture. No such controls figure in a prebiotic environment.
Characterization is critical. Without it, impurities accumulate. What prebiotic characterization might mean is anyone’s guess.
Given poor prebiotic reaction yields, it is impossible to envision a process in which the starting materials generate all of the desired products. We had to go back over and over again to generate molecular intermediates, a process known familiarly as “bringing up material from the rear.”
How would prebiotic chemistry bring up its own rear over and over again? It has kept no laboratory notebook to record the previous paths.
In our synthesis, we spent a great deal of time on separation and optimization. If byproducts are permitted to accumulate, they often will consume the new steps’ reagents and alter the course of the reaction.
This problem would plague abiogenesis, too.
It Stands to Reason
There must have been a chemical means, once upon a time, to generate an information-bearing molecule such as DNA or RNA. Since the 1960s, a number of biologists have suggested that the polymer is RNA rather than DNA. Such is the RNA World Hypothesis.39 And chemically activated ribonucleotides can polymerize to form RNA. So far so good.
But RNA is far less stable than DNA, and whatever the polymerization, it yields generic RNA, a molecule lacking sequence specificity. Had RNA researchers succeeded in producing a volume of random sentences—subtends flack lachrymose esurient—none of them would have imagined that they had succeeded in composing King Lear.
The coupling of a ribose with a nucleotide is the first step, and even those engrossed in prebiotic research have difficulty envisioning that process, especially for purines and pyrimidines.40 John Sutherland and his coworkers have proposed that pyrimidine ribonuceotides can form short sequences using arabinose amino-oxazoline and anhydronucleoside intermediates, all from simple compounds such as cyanamide, cyanoacetylene, glycolaldehyde, glyceraldehyde and inorganic phosphate. The use of inorganic phosphate changes the experiment’s basic conditions to a pH-buffered solution, thereby slowing decomposition pathways.41 But the work itself shows the intricacies required to generate the desired reactions.
The conditions were cleverly selected:
Although the issue of temporally separated supplies of glycolaldehyde [just-in-time (JIT) scenario 1] and glyceraldehyde [JIT scenario 2] remains a problem, a number of situations could have arisen [prebiotically, what and where?] that would result in the conditions of heating [careful control step 1 at 60°C ] and progressive dehydration [careful control step 2 by lyophilization which is water removal by ice sublimation under reduced pressure of <0.001 atmospheres] followed by cooling [careful control step 3 from 60°C to 23°C], rehydration [careful control step 4 with precise adjustments of pH] and ultraviolet irradiation [careful control step 5 with a selected 254nm light].42
There were also multiple purification steps [careful control of step 6], and ion exchanges using commercial resins [careful control of step 7]. All this for the synthesis of just one set of a mixture of adducts, and in racemic form.
It remains clear that the controlled conditions required to generate even a mixed set of select structures is painfully improbable.
Routes to each one of the requisite carbohydrates, lipids, nucleic acids, and proteins (polymers of amino acids) have been proposed in prebiotic studies. The attempted syntheses almost always create mixtures beset with the same difficulties.
From the data, the synthetic chemist can easily deduce that under prebiotic conditions the reaction in question is not likely to yield anything useful. With each added step, difficulties are compounded by improbabilities so overwhelming that no other field of science would depend upon such levels of faith.
Abiogenesis research would never be accepted in any other area of chemistry. The field is its own best enemy.
Extrapolation on Steroids
Sutherland and coworkers pointed out in 2015 that “[a] minimal cell can be thought of as comprising informational, compartment-forming and metabolic subsystems.”43 They also acknowledged that, to date, prebiotic chemistry has made ambitious extrapolations: “To imagine the abiotic assembly of such an overall [cellular] system [or subsystem], however, places great demands on hypothetical prebiotic chemistry.”44 Yet this revealing comment by Sutherland and his coworkers is coupled with their disclosure of a new experimental finding showing
that precursors of ribonucleotides, amino acids and lipids can all be derived by the reductive homologation of hydrogen cyanide and some of its derivatives … The key reaction steps are driven by ultraviolet light, use hydrogen sulfide as the reductant and can be accelerated by Cu(I)‑Cu(II) photoredox cycling.45
They assert boldly that, “all the cellular subsystems could have arisen simultaneously through common chemistry.”46 This has now raised the level of suppositions from mere molecule types to complex subsystems where molecules are working in concert toward a common functional goal. But compositions of a few molecule types, or even all of them, do not constitute a cellular subsystem. It is essential to emphasize that the authors only prepared precursors to the ribonucleotides, amino acids, and lipids, not the actual molecules, so the gross extrapolation is all the more disconcerting.
When reading the protocols for the suggested prebiotic-like precursors, one is struck by the high-level sophistication, expert synthetic prowess, and remarkable ingenuity of the researchers. Some reactions were run at room temperature, some at 60°C, others at 100°C and then washed with ice-cold water. Often the molecules prepared by these supposed prebiotic routes were not used, but had to be made more cleanly and in larger scale using purely synthetic methods and organic solvents, such as Lawesson’s reagent and tetrahydrofuran, respectively, “to simplify the handling procedures.”
JIT and precise order of addition protocols were used over and over again. One sees precise pH adjustments through the syntheses, use of ion exchange resins, and separations from the reaction mixtures because proceeding without separations would have destroyed the carefully prepared products. The preparation of cyanoacetylene on Cu(I) was suggested as a way to prepare it conveniently and store it for use when needed. CuCl was mixed with KCl to generate the Nieuwland catalyst, K[CuCl2], at 70°C. Then a separately generated source of acetylene gas was prepared from CaC2 and water. This gas was bubbled through the Nieuwland catalyst to prepare acrylonitrile (an unstable molecule that needs proper isolation and storage to inhibit its polymerization), which was then treated with KCN for 1h, then 5 equivalents of NH3 as a 13 molar NH3/NH4+ solution adjusted to pH 9.2 with NaOH to generate the desired aminopropionitrile.
All of the reactions were executed in separate clean vessels and properly isolated prior to proceeding to the next reaction.
This is just a sampling of preparations that are difficult even for the skilled synthetic chemist to execute. The routes afford very simple precursors to just a few of the many molecules within the building block class, and all their precursors were racemic if they even bore any possible stereoisomerism.
Dream On
The world’s best synthetic chemists, biochemists, and evolutionary biologists have combined forces to form a team—a dream team in two quite distinct senses of the word. Money is no object. They have at their disposal the most advanced analytical facilities, the complete scientific literature, synthetic and natural coupling agents, and all the reagents their hearts might desire. Carbohydrates, lipids, amino acids, and nucleic acids are stored in their laboratories in a state of 100% enantiomeric purity.
Would the dream team—please—assemble a living system?
Take your time, folks, take a few billion years.
Nothing? Well, well, well.
Let us assume that all the building blocks of life, and not just their precursors, have been made to a high degrees of purity, including homochirality where applicable—the carbohydrates, the amino acids, the nucleic acids, and the lipids. They are stored in cool caves, away from sunlight, and away from oxygen. These molecules are indifferent to environmental degradation.
And let us further assume that they are all stored in one comfortable corner of the earth, not separated by thousands of kilometers or on different planets.
And that they all exist not just in the same square kilometer, but in neighboring pools where they can conveniently and somehow selectively mix with each other as needed.
Now what? How does the dream team assemble them without enzymes?
Very well. Give the dream team polymerized forms: polypeptides, all the enzymes they desire, the polysaccharides, DNA and RNA in any sequence, cleanly assembled.
Ready now?
Apparently not.
We teach our students that when a mechanism does not support their observations, the mechanism must either be revised to support the facts or entirely discounted. They are not required to provide an alternative.
We are such stuff as dreams are made on. It has a ring among prebiotic chemists.
The Current State
Those who think scientists understand the issues of prebiotic chemistry are wholly misinformed. Nobody understands them. Maybe one day we will. But that day is far from today. It would be far more helpful (and hopeful) to expose students to the massive gaps in our understanding. They may find a firmer—and possibly a radically different—scientific theory.
The basis upon which we as scientists are relying is so shaky that we must openly state the situation for what it is: it is a mystery.
Images reprinted with permission from the American Chemical Society.
